
牛病毒性腹泻病毒SYBR Green荧光定量PCR方法的建立及其应用
2019-01-08 03:08崔尚金
现代畜牧兽医 2018年12期
刘 存 ,崔尚金 ⋆
(1.中国农业科学院北京畜牧兽医研究所,北京 100193;2.农业部兽用药物与诊断技术北京科学观测实验站,北京 100193)
牛病毒性腹泻-黏膜病(Bovine viral diarrhea-mucosal disease,BVD-MD)由牛病毒性腹泻病毒(Bovine viral diarrhea virus,BVDV)引起,主要以口腔、食管、胃、肠等部位黏膜侵蚀性或溃疡性病变、腹泻、白细胞减少、持续性感染及怀孕母牛流产或产畸形胎儿等症状为特征[1-2]。BVDV在世界范围内广泛流行,给养牛业国家造成严重的经济损失[3]。犊牛感染BVDV后,潜伏期7~9 d,发病率为2%~5%,犊牛死亡率可达90%。妊娠母牛在妊娠30~120 d间感染后可产持续性感染牛,持续性感染牛可持续排毒成为BVDV重要的传染源,是饲养场无法根除BVDV的重要原因之一。通过检测并淘汰持续性感染牛,是逐步实现BVDV净化的重要手段。
牛病毒性腹泻病毒(Bovine viral diarrhea virus,BVDV)是黄病毒科(Flaviridae)瘟病毒属(Pestivirus)成员,该病毒属还包括猪瘟病毒(Classical swine fever virus,CSFV)和羊边界病病毒(Border disease virus,BDV)[4]。BVDV基因组为单股正链RNA,由5’非翻译区(5′UTR)、ORF编码区、3’非翻译区(3′UTR)组成。BVDV基因组编码4种结构蛋白和8种非结构蛋白,分布依次为5’-Npro-capsid-Erns-E1-E2-P7-NS2/3-NS4a-NS4b-NS5a-NS5b-3’[5]。根据是否引起细胞病变,BVDV可分为两种生物型:致细胞病变型(Cytopathogenic,CP)和非致细胞病变型(Noncytopathogenic,NCP)[6];根据BVDV基因序列,BVDV又可分为BVDV-1和BVDV-2两个基因型。由于BVDV易突变且不同毒株间存在同源重组现象,因此BVDV基因亚型众多,目前BVDV-1分为1a~1u共21个亚型,BVDV-2分为2a~2d共4个亚型[7]。由于BVDV5’UTR高度保守,常作为靶标用于BVDV抗原的检测。由于NCP型BVDV不致培养细胞产生细胞病变,其病毒滴度测定常采用间接免疫荧光或者间接免疫过氧化物酶试验进行,耗时且不方便。荧光定量PCR方法是常用于病原检测以及定量的方法,且该方法特异性强、敏感性高、可重复性好、简便省时,可应用于BVDV培养过程中BVDV生长曲线的测定。本研究参考BVDV保守的5’UTR序列,设计引物建立BVDV RT-qPCR方法,为BVDV检测及培养过程一步生长曲线的测定奠定基础。
1 材料与方法
1.1 病毒与细胞牛病毒性腹泻病毒BVDV-BJ-2016株、牛副流感病毒3型BPIV3 Beijing株、牛传染性鼻气管炎病毒BHV-1 Beijing株均为作者所在实验室分离鉴定和保存,猪瘟病毒来源于市售弱毒活疫苗。经牛肾细胞(MDBK)传代培养BVDV,收取培养物-80℃保存备用。MDBK细胞由作者所在实验室保存。
1.2 主要试剂及仪器AxyPrep体液病毒DNA/RNA小量提取试剂盒为Axygen产品;PrimeScriptTMRT reagent Kit为TaKaRa公司产品;SanPrep柱式DNA胶回收试剂盒购于上海生工生物工程有限公司;EasyPure Plasmid MiniPrep Kit购于北京全式金生物技术有限公司;KAPA™SYBR®快速定量PCR试剂盒购于北京普凯瑞生物科技有限公司;7900HT Fast Real-time PCR仪为美国ABI公司产品。.4标准品DNA模板制备按照AxyPrep体液病毒DNA/RNA小量试剂盒操作说明书,从BVDV-BJ-2016株细胞培养上清中提取RNA。参照PrimeScriptTMRT reagent Kit说明书将提取的RNA反转录为cDNA,以获得的cDNA为模板进行标准品DNA模板的PCR扩增。标准品DNA模板扩增用引物:上游引物:5’-TAgCCATgCCCTTAgTAggAC-3’;下游引物:5’-ACTCCATgTgCCATgTACAgC-3’,片段大小约298 bp。PCR采用50 μL体系:上、下游引物各1 μL;cDNA模板 3 μL;PCR Mix 25 μL;ddH2O 20 μL。反应条件为:94℃5 min;94℃30 s,55℃30 s,72℃30 s,35个循环;72℃延伸10 min。PCR产物经琼脂糖凝胶电泳检测,回收预期目的条带与pMD18-T连接、转化,获得重组质粒pMD18T-5’UTR。用紫外分光光度计测定重组质粒的质量浓度,按照公式 copies/μL=(6.02×1023copies/mol)×(浓度g/mL)×10-3/(分子质量g/mol)计算拷贝数计算重组质粒拷贝数。
1.3 引物设计根据GenBank上登录的BVDV 5’UTR序列的保守序列,应用Primer Express 3.0设计特异性荧光定量RT-PCR引物,上游引物序列:5’-GGGNAGTCGTCARTGGTTCG-3’,下游引物序列:5’-TGTGCCATGTACAGCAGAGYTT-3’,目的片段大小为198 bp。上述引物由北京六合华大基因科技有限公司合成。
1
1.5 荧光定量PCR反应条件根据KAPA™SYBR®快速定量PCR试剂盒说明书,荧光定量PCR体系采用20 μL体系。以阳性标准品质粒为模板,采用矩阵法,对荧光定量PCR方法的退火温度(58、60、62、64℃)进行优化,并利用矩阵法对引物(10 μmol/L)使用量(0.4、0.6、0.8、1.0 μL)依次进行优化。
1.6 标准曲线的建立将重组质粒标准品进行10倍倍比稀释,选用 107、106、105、104、103、102、101copies/μL的重组质粒作为标准品为模板,以优化的反应条件进行荧光定量PCR检测,每个浓度标准品做3个重复。以log(拷贝数)为纵坐标、CT值为横坐标绘制标准曲线,并进行线性回归分析。
1.7 敏感性试验将制备的重组质粒标准品进行10倍梯度稀释,采用浓度为108、107、106、105、104、103、102、101、100copies/μL 的标准品为模板 ,应用建立的荧光定量PCR方法进行检测,确定其敏感性。
1.8 重复性试验采用浓度为108、107、106copies/μL的标准品质粒为模板进行3批重复性试验。每批次重复3次以检验其批内重复效果。在不同时间对该试验进行3次重复试验以检验该方法批间重复效果。
1.9 特异性试验应用建立的荧光定量PCR方法对BVDV-BJ-2016株、BPIV3 Beijing株、BHV-1 Beijing株以及CSFV的cDNA或DNA进行检测以评价所建立方法的特异性。
1.10 BVDV一步生长曲线测定将BVDV病毒液(BVDV-BJ-2016株)稀释1 000倍后接种MDBK细胞,在接毒后 2、4、6、8、12、24、36、48、60、72、84、96、108 h分别收集细胞培养上清1 mL,用于RNA的提取。RNA提取参照AxyPrep体液病毒DNA/RNA小量提取试剂盒说明书进行。提取的RNA用Nano Drop 2000测定质量浓度后,取1 μg RNA进行反转录,获得cDNA。取1 μL cDNA用本研究中所建立的SYBR Green荧光定量PCR方法进行检测,确定培养上清中BVDV基因组拷贝数的变化。根据所建立的标准曲线以及Ct值计算每毫升病毒液中所含BVDV基因组拷贝数,并以l g(copies/mL)为纵坐标、时间为横坐标绘制BVDV一步生长曲线。
2 结果与分析
2.1 BVDV 5’UTR RT-PCR扩增结果以提取的BVDV-BJ-2016株cDNA为模板,采用BVDV 5’UTR的特异性引物进行RT-PCR扩增,经琼脂糖凝胶电泳检测,目的片段约298 bp,符合预期目的条带大小。回收目的条带,连接于pMD18-T载体,以构建阳性模板。将鉴定为阳性的重组载体命名为pMD-5’UTR。
2.2 荧光定量PCR方法条件优化根据KAPA™SYBR®快速定量PCR试剂盒说明书确定反应体系为20 μL,经过优化最终确定为qPCR Mix 10 μL,上、下游引物(10 μm/L)0.6 μL,Rox 0.4 μL,模板 1 μL,ddH2O补至20 μL。经过对反应条件的优化,最终确定该方法最佳的反应条件为:95℃预变性1 min;95℃变性15 s,60℃退火30 s(收集荧光),共计40个循环。
2.3 标准曲线的建立将重组质粒pMD-5’UTR经10 倍梯度稀释,分别以 107、106、105、104、103、102、101copies/μL的重组质粒作为标准品模板,进行荧光定量PCR检测获得扩增曲线(见图1B)。以起始模板浓度的对数为横坐标、Ct值为纵坐标绘制标准曲线。通过熔解曲线分析(见图1A),可见标准样品均出现了单一的峰,表明该方法为特异性扩增。标准曲线方程为y=-3.5197+40.4431,相关系数R2=0.9973,各浓度标准品间具有良好的线性关系。
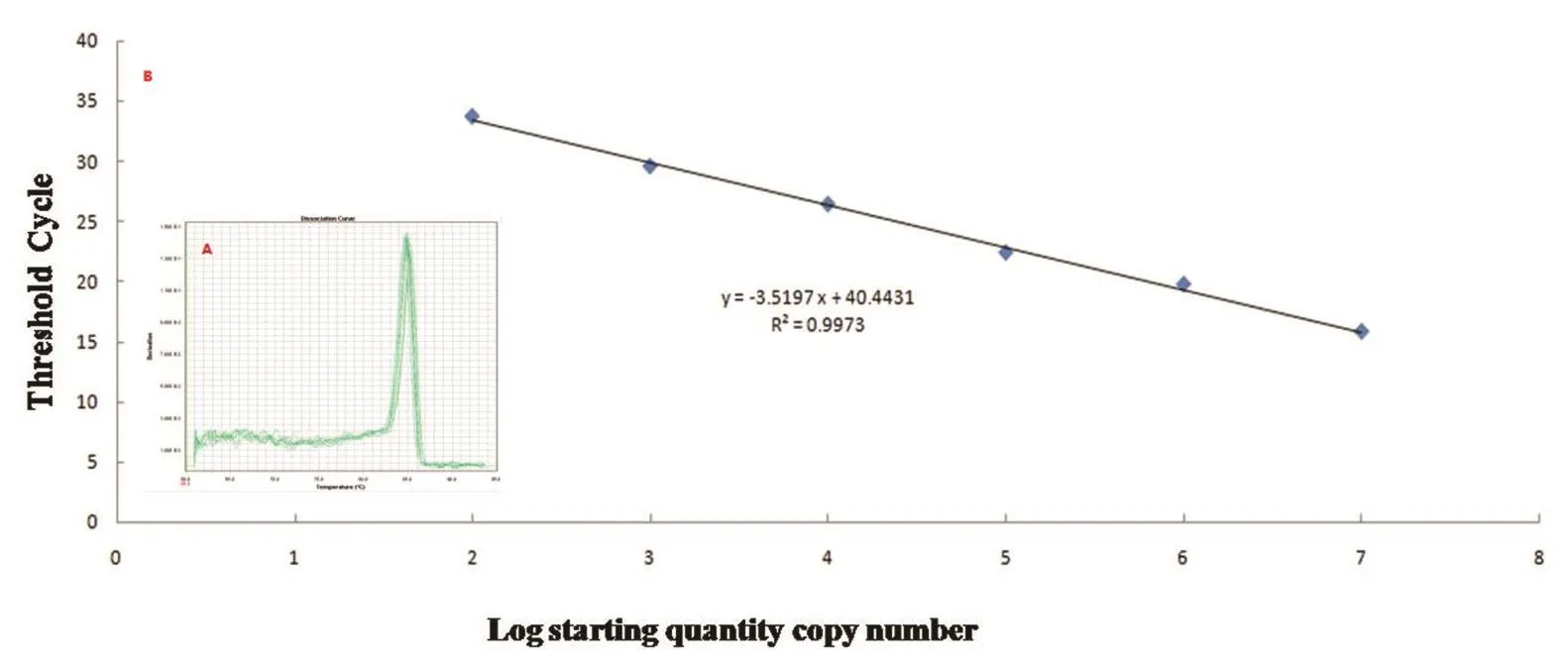
图1 BVDV RT-qPCR标准曲线Fig.1Standard curve of BVDV RT-qPCR
2.4 敏感性试验 分别以107、106、105、104、103、102、101、100copies/μL的重组质粒作为标准品模板利用所建立的荧光定量PCR方法进行检测,检测结果均呈现典型的S型曲线(图2)。该建立的方法最低检测限为10.0 copies/μL。
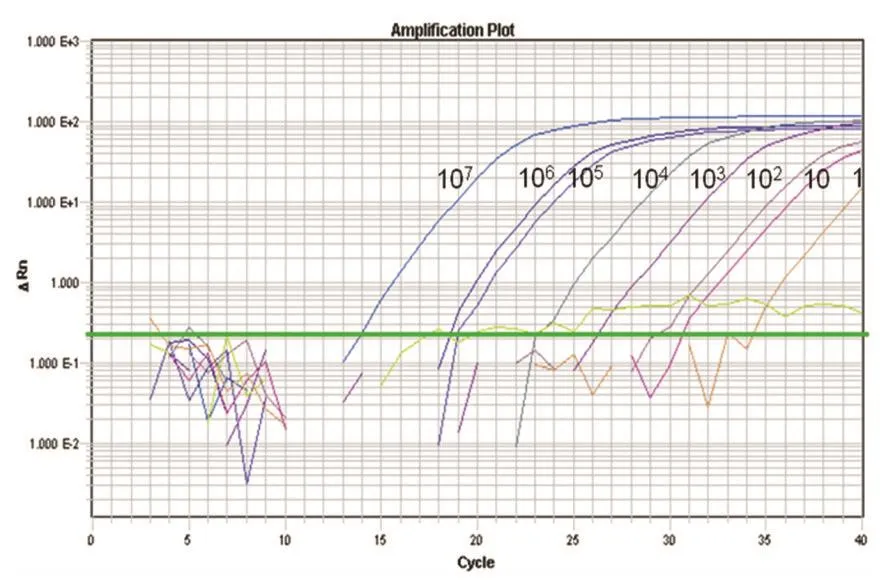
图2 BVDV RT-qPCR方法敏感性试验结果Fig.2Result of sensitivity test of BVDV RT-qPCR
2.5 特异性试验利用建立的SYBR Green荧光定量PCR方法对BVDV、BPIV3、BHV-1、CSFV的核酸进行检测,结果显示BVDV和CSFV样品为阳性,其他病毒样品均为阴性。该结果表明,猪瘟病毒对本研究中所建立的RT-qPCR方法存在干扰(图3),即所建立的SYBR Green荧光定量PCR方法不能够区别鉴定BVDV与猪瘟病毒,这可能由于BVDV与猪瘟病毒同属瘟病毒属,而在瘟病毒属成员中基因组5’非编码序列相对保守。特异性试验结果显示,所建立的SYBR Green荧光定量PCR方法虽不能特异性地鉴别BVDV与猪瘟病毒,但其与牛副流感病毒3型、牛传染性鼻气管炎病毒并不存在交叉反应,可应用于瘟病毒与其他不同病毒的诊断中。
2.6 重复性试验选取浓度为108、107、106copies/μL的标准品质粒为模板,分别进行3批批内和批间重复试验以评价所建立SYBR Green荧光定量PCR方法的重复性。结果显示,所建立的荧光定量PCR方法的批内和批间重复试验的变异系数均小于5%,说明所建立SYBR Green荧光定量PCR方法具有良好的重复性。
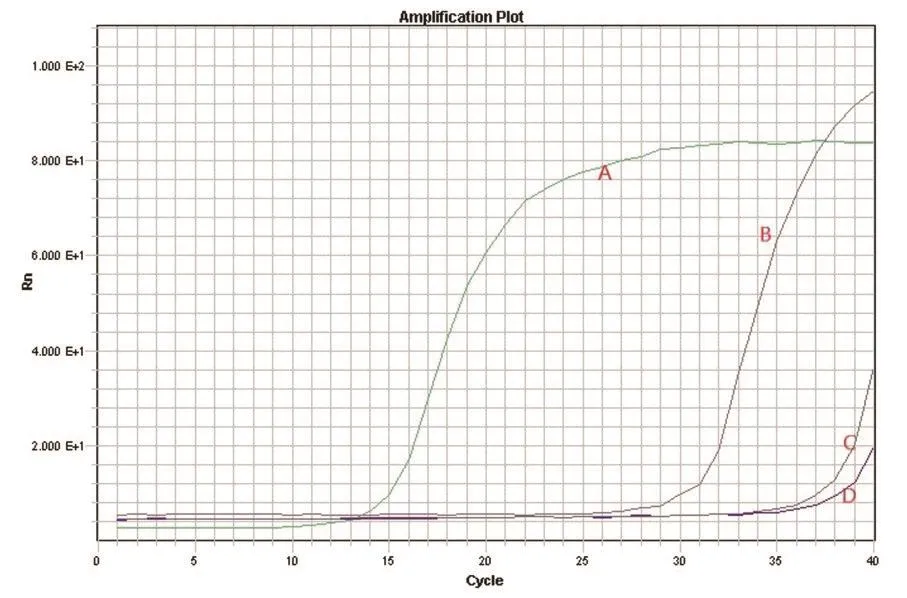
图3 BVDV RT-qPCR特异性试验Fig.3Specificity test of BVDV RT-qPCR
2.7 BVDV一步生长曲线的测定在BVDV接种MDBK细胞后,收取接毒后2、4、6、8、12、24、36、48、60、72、84、96、108 h的细胞培养上清。收取上清用所建立的RT-qPCR方法进行检测,获得Ct值,将Ct值代入标准方程后计算拷贝数。以拷贝数的对数为纵坐标,以样品收集时间为横坐标绘制BVDV一步生长曲线(图4)。BVDV一步生长曲线显示,在接毒后12~48 h间为BVDV对数生长期。在接毒48 h以后,BVDV基本处于稳定期。从绘制的BVDV一步生长曲线提供的参考,BVDV在接毒后72~84 h间收获病毒即可。
3 讨论
牛病毒性腹泻病毒感染是危害养牛业的重要疾病,开发检测和诊断牛病毒性腹泻的快速诊断技术对牛病毒性腹泻的防控及净化、推动养牛业健康发展具有重要的意义。为了预防与控制牛病毒性腹泻病毒感染,科研人员建立了诸多分子生物学诊断方法。应用地高辛、生物素等标记物标记核酸探针,建立核酸杂交检测方法,快速、简便、经济地检测BVDV[8]。白泉阳等[10]、宋爽等[11]针对BVDV保守的5’UTR设计引物和探针,建立Taqman荧光定量PCR方法,方便、快速地检测BVDV,该方法敏感度高、重复性好。
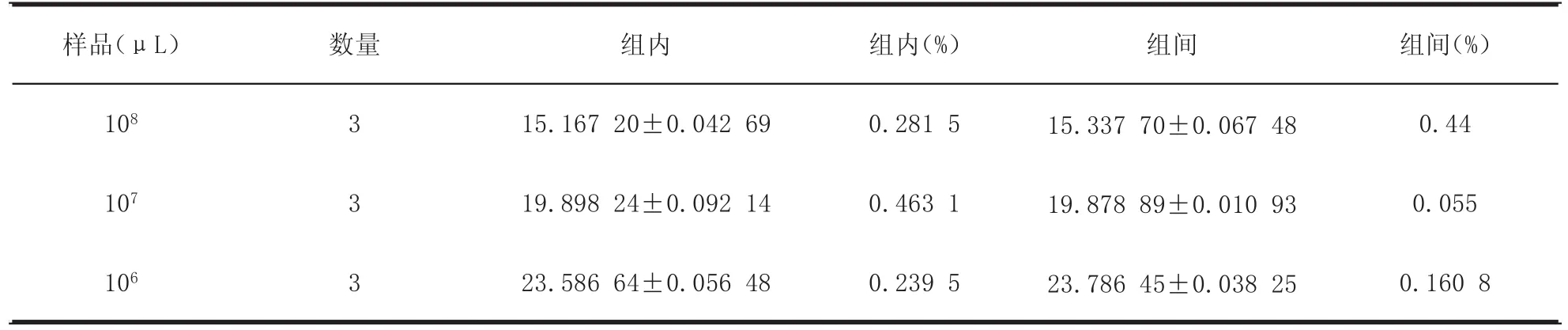
表1 BVDV RT-qPCR重复试验结果Table 1Inter-assay and intra-assay reproducibility test of the RT-qPCR
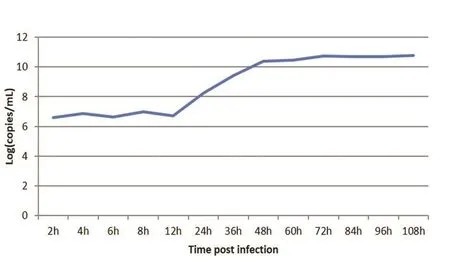
图 4 BVDV在MDBK细胞上一步生长曲线Fig.4 One-step growth curve of BVDV in MDBK cells
荧光定量PCR方法具有操作简便、特异性高、敏感性高、重复性好、可实时定量等特点。实时荧光定量PCR包括探针法和染料法,其中染料法相对探针法价格更低。目前荧光定量PCR已经成为实验室快速检测和诊断病原的重要手段[12]。对于非致细胞病变型牛病毒性腹泻病毒在培养细胞上不产生细胞病变,通过间接免疫荧光试验或间接免疫过氧化物酶试验测定接毒后不同时间点收获病毒的TCID50,进而绘制非致细胞病变型牛病毒性腹泻病毒在培养细胞上的生长动力学曲线,此种测定方法耗力费时。而应用荧光定量PCR方法可快速定量地对病毒基因组进行检测,因此本试验将荧光定量PCR方法应用于BVDV一步生长曲线的绘制中。
为了实现测定BVDV NCP型一步生长曲线,本试验根据牛病毒性腹泻病毒5’UTR保守序列设计荧光定量PCR引物,建立了检测牛病毒性腹泻病毒的SYBR Green荧光定量PCR方法并将该方法应用于BVDV一步生长曲线的测定中。研究中所建立的SYBR Green荧光定量PCR方法具有良好的重复性和敏感性,但其存在不能特异性鉴别BVDV与猪瘟病毒的缺陷。这一缺陷,可根据瘟病毒属成员5’UTR序列中的差异序列区设计特异性的探针,以改善荧光定量PCR方法的特异性。该方法的建立和应用,为研究非致细胞病变型BVDV分离毒株的生长特性的研究提供了一定的参考。
猜你喜欢
广东药科大学学报(2022年3期)2023-01-04
生物学通报(2022年1期)2022-11-22
湖南饲料(2021年4期)2021-10-13
世界科学技术-中医药现代化(2020年2期)2020-07-25
中成药(2018年12期)2018-12-29
江西医药(2018年8期)2018-10-24
中国医药指南(2017年3期)2017-11-13
中成药(2017年6期)2017-06-13
医学研究杂志(2015年4期)2015-06-10
中国民族民间医药·下半月(2014年2期)2014-09-26
