
N-羟乙酰神经氨酸分子的结构性质
2018-12-29 08:30梁美莲周樱子李洪英徐阿奇晏印雪刘春丽曾雪峰朱秋劲
食品科学 2018年23期
常 瑞,梁美莲,周樱子,李洪英,徐阿奇,晏印雪,刘春丽,曾雪峰,朱秋劲*
(贵州大学酿酒与食品工程学院,贵州省农畜产品贮藏与加工重点实验室,贵州 贵阳 550025)
唾液酸是含9 个碳原子并具有吡喃糖结构的酸性氨基糖,系统命名为5-氨基-3,5-二脱氧-D-甘油-D-半乳糖壬酮糖。N-羟乙酰神经氨酸(N-glycolylneuraminic acid,Neu5Gc)是其中重要的一种,其余两种分别是3-脱氧-D-甘油-D-半乳壬酮糖(3-deoxy-D-glycerol-D-galactone ketone,KDN)和N-乙酰神经氨酸(N-acetylneuraminic acid,Neu5Ac)[1]。由于人类进化过程中编码表达胞苷单磷酸N-乙酰神经氨酸羟化酶的基因发生了突变,因此人体不能合成Neu5Gc[2]。正常人体中存在的唾液酸只有Neu5Ac,母乳中质量浓度约为0.3~1.5 mg/mL,其是促进人脑发育的营养物质[3]。Neu5Gc在红肉等加工肉制品中有微量存在[4],流行病学研究表明饮食摄入的Neu5Gc和炎症的产生有一定联系[5]。2015年国际癌症研究机构基于结/直肠癌的流行病学结果,将加工肉制品列为I类致癌食品[6]。
分子的结构参数与其物理化学反应活性有密切的关系,量子化学计算是研究分子结构参数的有力工具。Djeradi等通过对黄酮化合物福井描述符的计算,构建了可预测抗氧化活性的定量构效关系模型,模型和实验值的相关系数达0.815 9[7]。不同加工方法在可一定程度上降低食品中Neu5Gc水平[8]。已报道的有物理法(高温油炸、蒸煮)、生物化学法(发酵、酶制剂)等,这些方法降低效果并不理想。食品加工过程中的溶剂对食品组分有一定程度影响[9],考察加工过程常用的脂溶性溶剂和有机酸类对Neu5Gc等食品危害因子的反应活性可为提高其解离效果提供一定参考。
目前关于唾液酸分子结构的理论研究主要集中在Neu5Ac不同构象。Veluraja等用半经验算法研究了唾液酸化的低聚糖的优势构象及其与唾液酸酶的结合[10]。Sawada等在B3LYP/6-31G(d,p)理论水平讨论了Neu5Ac的最佳构象和水分子相互作用时的分子间氢键[11]。van Lenthe等采用HF/6-31G(d)理论水平研究了6 种唾液酸衍生物的电荷、轨道信息以及与唾液酸相关的酶类[12]。Priyadarzini等研究了Neu5Ac在生物环境中的三维构象和分子间氢键作用[13]。但关于Neu5Gc的分子结构和物理化学性质尚鲜有研究。因此,本实验通过量子化学密度泛函理论研究Neu5Gc分子电子结构、不同溶剂环境下分子反应活性和相关物理化学性质,以期为研究其食品加工环境下的反应活性和有效解离提供理论参考。
1 材料与方法
1.1 Neu5Gc分子电子结构分析
Neu5Gc的分子结构取自PubChem数据库,在M062X/6-31+G(d,p)水平上完成了对Neu5Gc分子的几何构型优化和频率振动分析,得到能量极小的无虚频结构。在优化后的结构上利用Multiwfn3.5程序进行分子表面静电势、前线分子轨道、自然电荷分布、概念密度泛函活性指数的计算。
概念密度泛函是密度泛函理论的化学活性理论,包括全局描述符(电负性Χ、化学势μ、化学硬度η、化学软度S、亲电指数ω)和局部描述符(福井函数等)。分子轨道的分布在一定程度上可以反映分子的反应活性。硬软酸碱理论(hard-soft-acid-base,HSAB)认为,缺乏电子的亲电试剂可以看作路易斯酸,富电子的底物可以看作路易斯碱,较硬(软)的路易斯酸容易与较硬(软)的路易斯碱相互作用。Parr等证明了用密度泛函计算分子化学势时与元素的电负性等同[14]。全局描述符可由前线分子轨道分布近似得到,计算公式如式(1)~(5)[15]。
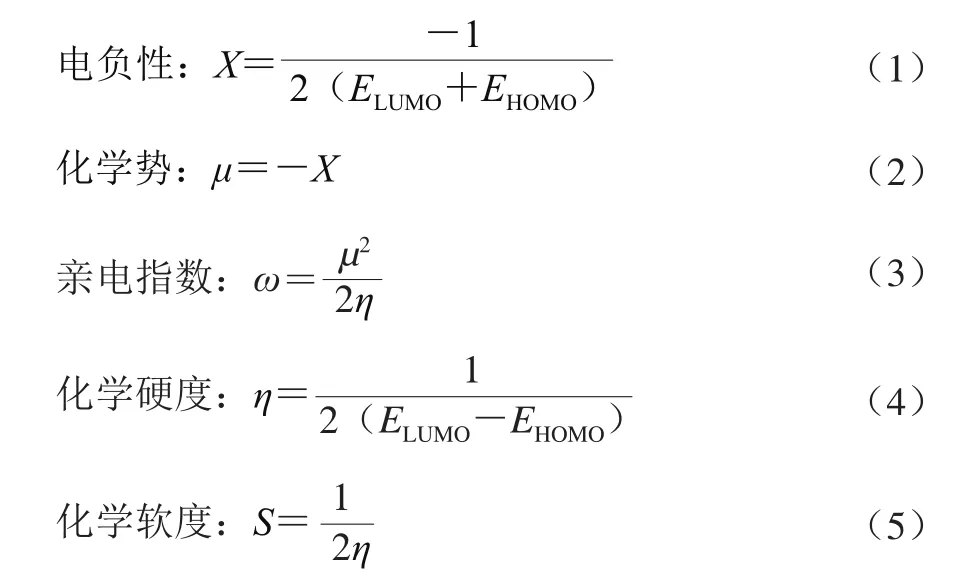
式中:ELUMO为最高低据轨道能级;EHOMO为最高占据轨道能级。
福井函数可反映体系电子数变化时各个位置上电子密度的变化程度,是重要的局部活性描述符。福井函数越大的位点,其相应的反应活性也越大。在轨道弛豫效应不显著的情况下,前线轨道理论是对福井函数的一个合理的近似,可由公式(6)~(8)计算[16]。

式中:ρN(r)、ρN+1(r)、ρN-1(r)分别表示体系在初始态、电离掉一个电子和结合一个电子的状态时的电子密度;ρHOMO(r)、ρLUMO(r)分别为分子最高占据轨道和最低非占据轨道的电子密度。
将福井函数收缩到原子上就得到简缩型福井函数(condensed Fukui function,CFF),可实现对原子电荷进行定量分析。此处用qA表示分子中原子A的Hirshfeld电荷,计算表达式如式(9)~(11)[17]。
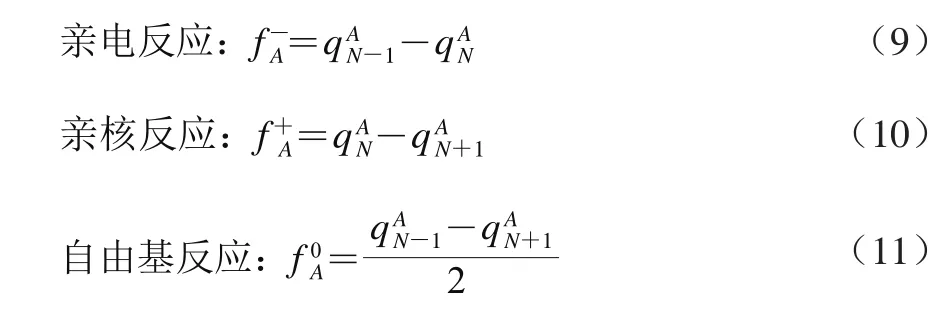
根据Neu5Gc分子结构含多个羟基的特点,为分析其分子内弱相互作用,在M062X-D3/6-311+G(d,p)水平气相条件下优化结构并产生波函数文件。根据Johnson等[18]提出的约化密度梯度函数(reduced-density gradient,RDG)理论,进行约化密度梯度函数等值面分析;为表征弱相互作用的关键临界点,根据分子中原子(atom in molecular,AIM)理论进行电子密度拓扑分析[19]。
1.2 Neu5Gc化学常数的计算
通过对有机反应过程定量计算,可以得到一些关键物理化学参数。在M062X/6-31+G(d,p)水平气相优化后的基础上,采用相应的理论基组水平计算Neu5Gc的解离常数、分配系数、键解离能。
1.2.1 Neu5Gc解离常数的计算
化合物的酸碱解离常数pKa值是重要的分子参数之一。Neu5Gc分子含一个羧基基团,在水溶液中可以发生解离呈现酸性。已知Neu5Ac的解离常数pKa为2.6。且羧酸化合物(HA)在水溶液存在以下热力学循环方程[20]。

式中:△G(aq)为水溶液下质子解离过程的自由能;△G(g)为气相下质子解离过程自由能;△Gsolve为溶解自由能。
张霞[21]提出了利用气相条件下相对吉布斯自由能差法计算pKa值的简便算法。Neu5Gc、Neu5Ac及其阴离子在气相条件下的吉布斯自由能以热力学高精度组合算法CBS-QB3进行。其中,△G(HA)是中性和阴离子态下相对吉布斯自由能差,其提出的计算表达式如式(15)所示。
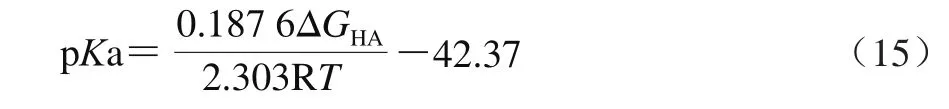
1.2.2 Neu5Gc分配系数的计算
有机物在正辛醇-水中的分配系数lg Poct/wat可以表示其对水相和醇相体系的亲和性差异,可以一定程度上模拟体内细胞膜和细胞溶质的环境。Ho等[22]认为,隐式溶剂模型下,溶解自由能为溶剂模型下单点能和气相条件下单点能的差值。根据Bannan等[23]提出的公式,正辛醇-水两相体系下分配系数的计算如式(16)、(17)所示。

式中:E辛醇和E水分别代表化合物在相应溶剂下的单点能。隐式溶剂模型不同于显示溶剂模型,它将溶剂环境认为是连续而非离散的状态,更加趋近于真实体系,故采用SMD(solvation model density)隐式溶剂模型计算,计算水平为M05-2X/6-31G(d)。根据Neu5Gc结构相似性,选择已知油水分配系数的β-D-葡萄糖进行对比计算。
1.2.3 Neu5Gc不同羟基位点的脱氢解离能
键解离能(bond dissociation energy,BDE)是破坏某一化学键所需要的能量。常用于表征黄酮类、多酚类的抗氧化性,Neu5Gc是吡喃糖衍生物,根据糖类化合物的羟基和清除自由基活性有一定相关性[24],Neu5Gc不同部位羟基的脱氢解离能计算公式如式(18)所示[25]。

式中:H(MO·)代表脱氢后的自由基片段的焓值;H(MOH)代表母体分子焓值。
溶剂化效应是指溶剂与溶质发生作用时会在溶质周围形成厚度不同的溶剂壳。介电常数常用于反映溶剂的极性,溶剂溶解度参数和极性对分子构型和电荷分布有一定影响。为考虑不同溶剂对解离能的影响,选取不同介电常数的溶剂(苯ε=2.27、四氯化碳ε=2.24、乙酸ε=6.25、乙醇ε=24.85、乳酸ε=22.00、甲酸ε=51.10、水ε=78.35)在SMD溶剂模型下进行计算。其中,溶剂乳酸根据Gaussian 16手册进行自定义溶剂参数拟合。优化水平为M062X/6-31+G(d,p),单点能在def2TZVP基组下进行。计算热力学校正量时,采用零点能校正因子0.967。
1.3 Neu5Gc红外和紫外光谱的模拟
在M062X/6-31+G(d,p)水平气相优化的结构基础上进行理论红外光谱模拟。采用的频率校正因子为0.94。为比较不同溶剂条件下Neu5Gc的理论紫外光谱,根据含时密度泛函理论在PBE1PBE/6-311G(d)水平下优化结构,TZVP基组下拟合光谱[26]。本实验中所有的量子化学计算部分在贵州大学云计算平台Gaussian 16软件包上完成,波函数分析部分采用开源的Multiwfn 3.5程序完成[27]。
2 结果与分析
2.1 Neu5Gc分子电子结构
在Gaussian 16程序中搭建好Neu5Gc的分子结构模型。M062X/6-31+G(d,p)水平下同时进行几何构型优化和振动分析,得到无虚频的能量极小结构(图1A)。对几何构型优化产生的波函数文件进行Multiwfn 3.5程序分析,得到能够描述电荷分布和静电相互作用的分子表面静电势分布图(图1B)。以气相条件下为例,Neu5Gc最高占据轨道能级(EHOMO)和最低空轨道能级(ELUMO)分布如图2所示。Neu5Gc在不同溶剂下的EHOMO、ELUMO及全局活性指数如表1所示。Neu5Gc不同羟基氧原子上的简缩型福井函数值如表2所示。
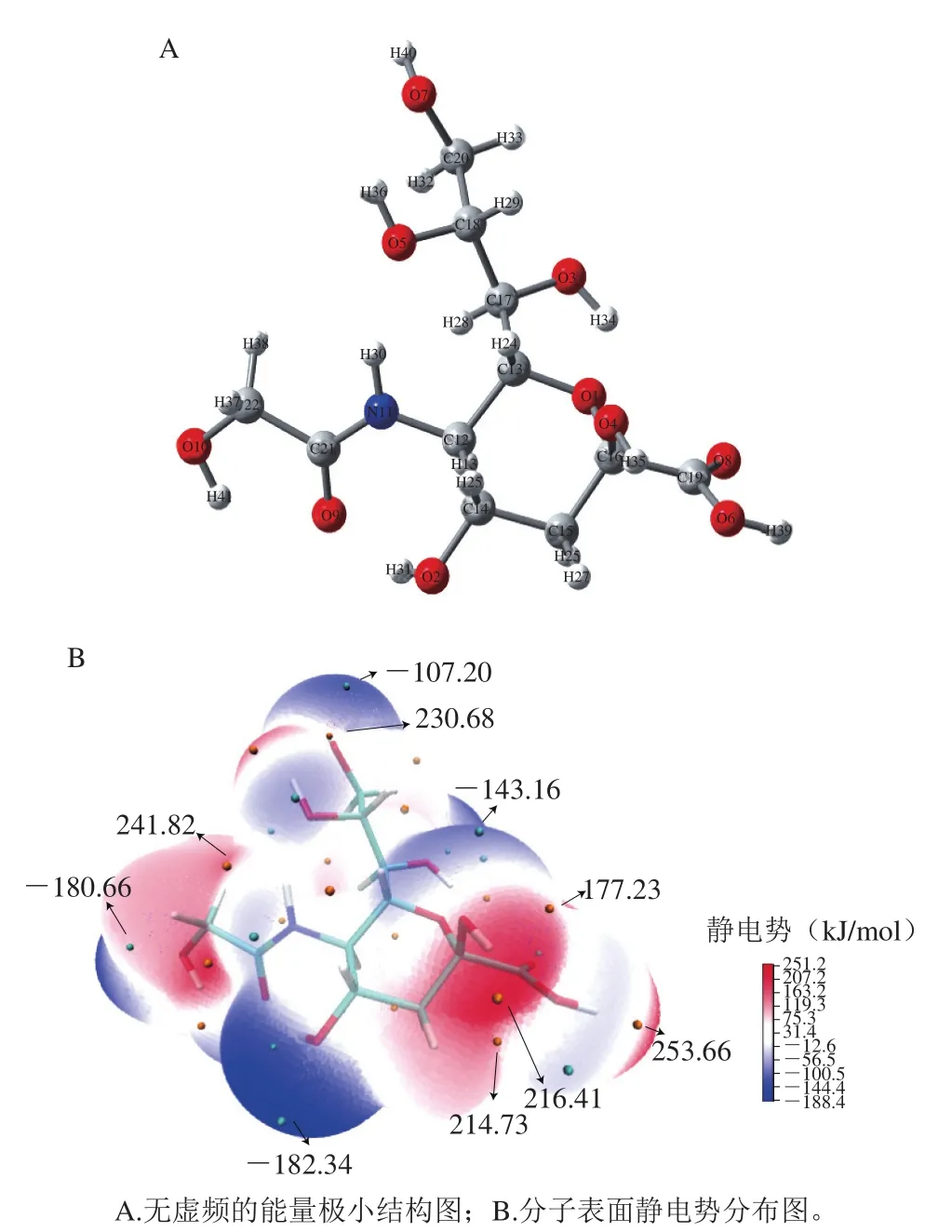
图1 Neu5Gc优化后构型及分子表面静电势分布Fig.1 Optimized geometry and electrostatic potential of Neu5Gc
从图1A可看出,优化后的Neu5Gc分子吡喃环上的取代基团均处于能量较低的平伏键位置,这表明优化过程是合理的。分子表面静电图上颜色深浅反映静电势的强弱,正的静电势区域用红色表示,负的区域用蓝色表示。由图1B可看出,静电势在负相区域的极小值为-182.34 kJ/mol,位于O2位附近,其次为O10位附近(-180.66 kJ/mol),表明这些区域负电荷集中,亲电活性较高。静电势在正相区域的极大值是253.66 kJ/mol,位于羧基基团O6附近,其次为C14位附近(214.73 kJ/mol)、C15位附近(216.41 kJ/mol),表明这些区域正电荷集中,亲核性较高。Garzón等对香豆酸与自由基攻击位点的研究也表明,羟基和羧基易受到较高亲核能力的自由基攻击[28]。唾液酸家族的燕窝酸作为糖类衍生物[29],其抗氧化活性可能也和其羟基、羧基有一定关联。

图 2 Neu5Gc HOMO和LUMO轨道分布图Fig.2 HOMO and LUMO orbital map of Neu5Gc molecules
根据前线分子轨道理论,EHOMO表征分子给电子的能力,EHOMO越大,给电子能力越强。ELUMO表征分子吸电子的能力,ELUMO越小,吸电子能力越强。前线分子轨道能级差△E能垒值越小,则表征分子中电子越容易发生跃迁,反应活性就越强[30]。用红色和绿色分别代表轨道的正负相位,结合图1A、2A可以看出,Neu5Gc的HOMO轨道主要分布在酰胺基团(N11—C21=O9)及其附近,表明该区域易受到亲电试剂攻击。从图2B可以看出,LUMO轨道主要分布在羧基基团(O6—C19=O8)处,表明易受到亲核试剂攻击,这和分子表面静电势的分析是一致的。Zhao Gang等对胶原蛋白氨基酸的轨道分析也显示出HOMO轨道主要集中在氨基区域,LUMO轨道主要集中在羧基区域[31]。
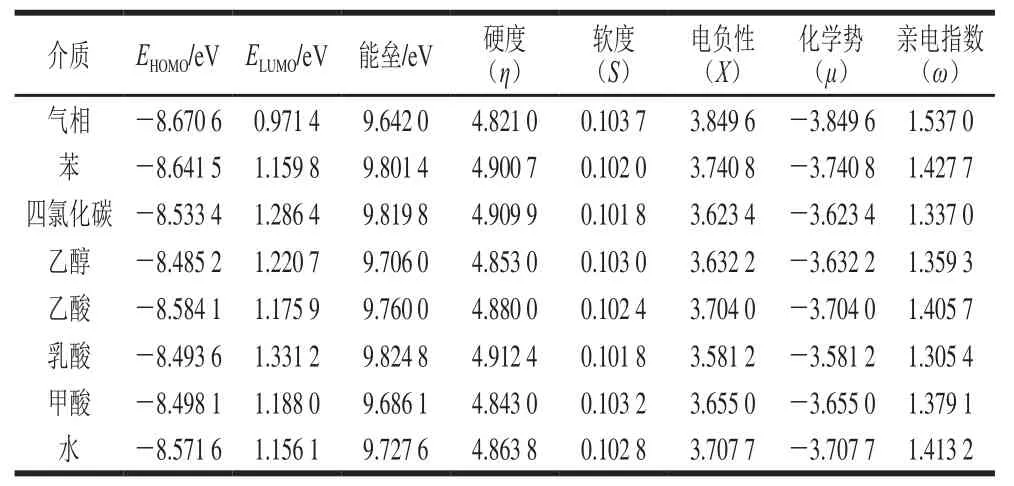
表1 不同溶剂下Neu5Gc全局活性参数M062X/6-31G(d,p)Table1 Neu5Gc global activity parameters at M062X/6-31G(d,p) level in different solvent environments
从表1可以看出,EHOMO最大的是乙醇条件下(-8.485 2 eV),最小的是气相条件下(-8.670 6 eV)。表明在乙醇条件下,Neu5Gc比较容易失去电子,还原性强。ELUMO最大的是乳酸条件下(1.331 2 eV),气相条件下则最小(0.971 3 eV),这表明乳酸条件下Neu5Gc吸电子能力较弱。轨道能级差气相条件下最小(9.642 0 eV),在乳酸条件下最大(9.824 8 eV),表明其在气相条件下不稳定。
由表1可知,Neu5Gc在气相条件下具有最低的化学硬度,最高的化学软度;乳酸条件下则有最高的化学硬度,最低的化学软度。乙酸和甲酸下的化学硬度也较气相条件下强。表明Neu5Gc可作为较硬的路易斯碱,易于和乳酸等路易斯酸发生反应。Chen Yue等对食品中Neu5Gc含量检测发现,酸奶、奶酪等发酵乳制品中的含量明显低于其他动物性食品[32]。从表1中还可看出,乳酸条件下Neu5Gc的电负性最低,亲电指数最小,化学势最高,这表明乳酸不仅可以提高食品抗氧化性[33],还可以与黄曲霉毒素等危害因子进行相互作用达到降解的效果[34]。
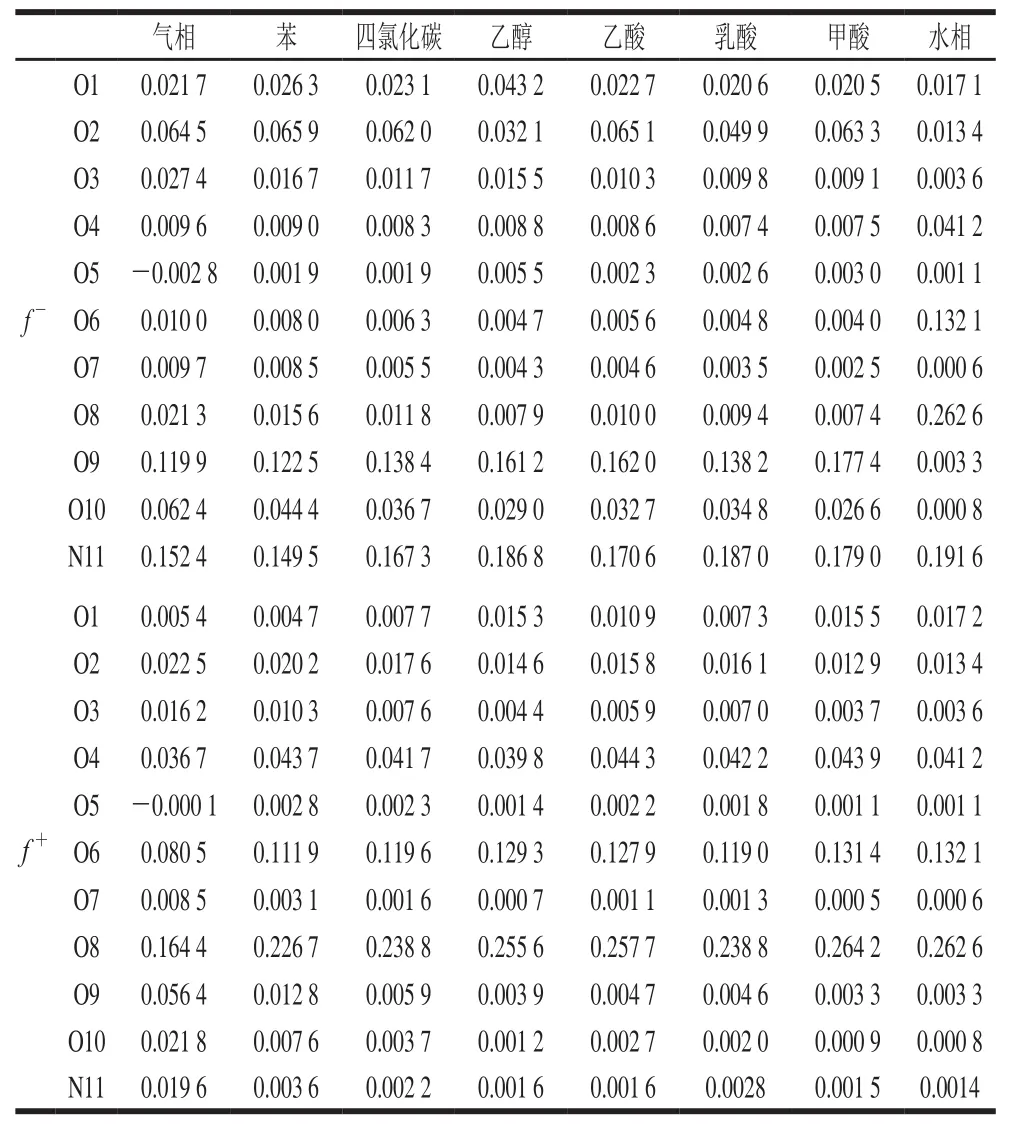
表2 Neu5Gc氮氧原子简缩型福井函数值Table2 Neu5Gc nitrogen oxygen atom condensed Fukui function values
简缩型福井函数能够对分子中的原子定量分析其亲电亲核反应活性,f-表征亲电活性,f+表征亲核活性[35]。已知氮氧原子的电负性大于碳原子,故此处分析Neu5Gc的氮氧原子。从表2可以看出,在以上8 种条件下,Neu5Gc中氮原子的f-值均大于氧原子的值,且在水相下最大。f-值最低为气相条件下O5位,最大为水相条件下O8位,O2、O9、O10位数值也略高于其他氧原子,这和HOMO对亲电位点的分析是一致的。羧基部位O4、O6、O8位的f+值总体大于其他氧原子,且f+值最大为甲酸条件下O8位,这和LUMO亲核活性分析结论一致。
分子体系内的相互作用范围可以是在原子核附近、化学键附近、弱相互区域、分子边缘。约化密度梯度函数基于电子密度,能够对局部的非共价相互作用进行定义。AIM理论的电子密度拓扑分析可以显示电子密度梯度为零的键临界点,临界点处的电子密度和键的强度有关[36]。Neu5Gc分子内弱相互作用RDG等值面和AIM电子密度拓扑分析结果如图3所示。图3A中蓝色部分主要是强的吸引作用,如氢键和卤键等;绿色的部分主要是范德华力,偏红色的部分主要为较强的斥力作用,如空间位阻效应。图3B中蓝色的点表示键临界点,红色表示环临界点。从图3A可看出,Neu5Gc分子吡喃环内和羟基氧上主要是位阻效应和弱的范德华力,乙酰基团和邻近羟基产生了一定的位阻效应。受乙酰基氮原子、邻近羟基氧原子电负性影响,氮原子上氢原子和O5产生了氢键作用。由图3B可看出,电子密度拓扑分析的结果和RDG分析一致,键临界点分布在RDG等值面上的强静电吸引力处,环临界点分布在空间位阻处。通过对键临界点连接的原子的距离分析发现,其均在氢键形成的键长范围内。综上分析,可得出Neu5Gc分子内形成的氢键分布在O5—H30、O9—H31、O1—H34处,相应的距离为1.98、1.86、1.90 Å。由于Neu5Gc分子内可形成3 个较强的氢键,这可能是导致其在食品热加工过程中稳定不易解离的原因。梁美莲等用高温蒸煮和油炸处理牛肉,发现其中Neu5Gc含量并没有明显降低[37]。
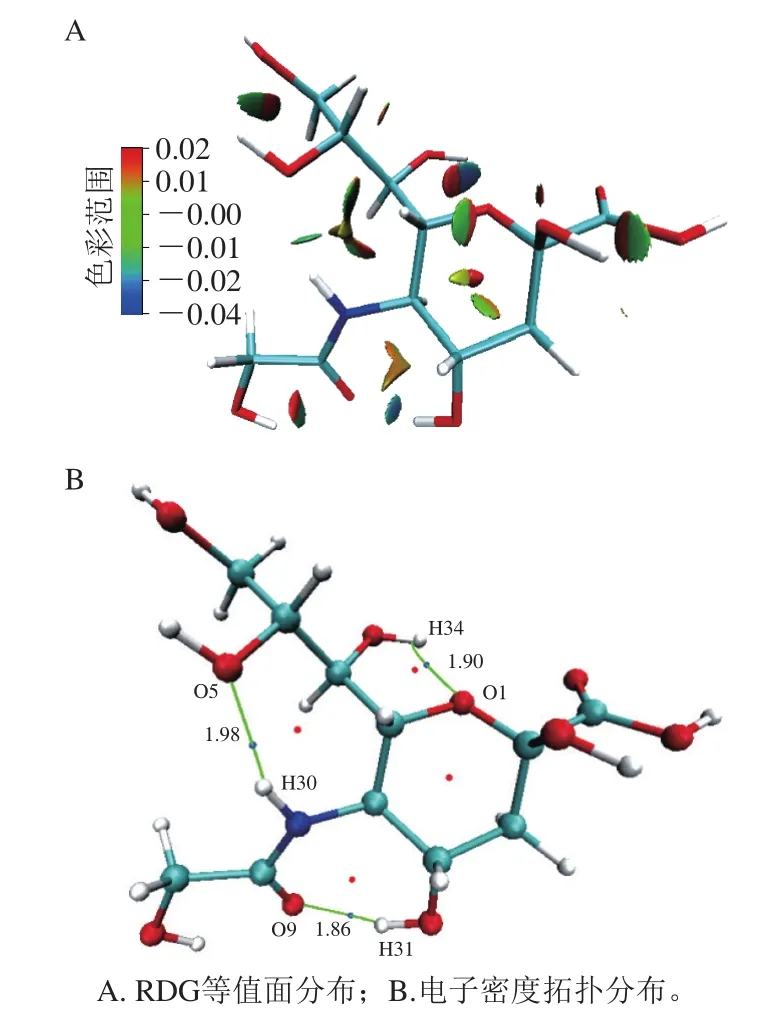
图3 Neu5Gc RDG等值面与电子密度拓扑分布Fig.3 RDG isosurface and electron density topological distribution of Neu5Gc
自然电荷分布是表征分子内不同原子上所带电荷大小的参数。电荷的分布也可以反映分子体系的亲电、亲核活性,负电荷越集中的区域其亲电活性也强[38]。计算得到的Neu5Gc氮氧原子的自然电荷分布如表3所示,气相条件下O5位电荷最小,O7位次之;溶液条件下,电荷大小为结果O7位<O5位、O9位<O8位,表明亲电活性O7位>O5位、O9位>O8位,这和f-值是一致的。O6位和N11在气相条件下电荷最小,其余氧原子在甲酸条件下电荷最小,乙酰基(氧原子O9)和羧基氧(原子O8)则在气相条件下电荷最大,表明亲电活性随溶剂有一定差异性。对羟基氧原子O3、O4、O5、O7、O10位,不同溶剂下电荷满足四氯化碳<苯<水相<乙醇,表明其在非极性溶剂下易发生亲电反应;不同有机酸下电荷大小满足甲酸<乳酸,且二者电荷小于气相,表明酸性条件下Neu5Gc分子活性较气相条件下增强。

表3 Neu5Gc氮氧原子自然电荷分布Table3 Natural charge distribution of nitrogen and oxygen atoms of Neu5Gc
2.2 Neu5Gc化学常数的计算
2.2.1 解离常数、分配系数的计算
解离常数和油水分配系数与食品的风味释放、包材迁移、生物利用度、代谢转化等有关。Neu5Ac和Neu5Gc水溶液中的pKa值计算结果如表4所示。对Neu5Ac、Neu5Gc和β-D-葡萄糖在正辛醇和水相下的单点能进行分配系数计算,结果如表4所示。计算得到Neu5Ac的pKa为2.20,与已知的实验值2.60相比,绝对误差为0.4,由此计算得到Neu5Gc的pKa值为1.84。对唾液酸母核结构类似物β-D-葡萄糖的分配系数计算值为-3.70,与已知的实验值相比,绝对误差为0.46。计算得到Neu5Gc的lg Poct/wat为-3.85,表明其具有良好的亲水性,其比Neu5Ac的-3.41稍低,这可能是由于Neu5Gc是Neu5Ac乙酰基羟化的产物。
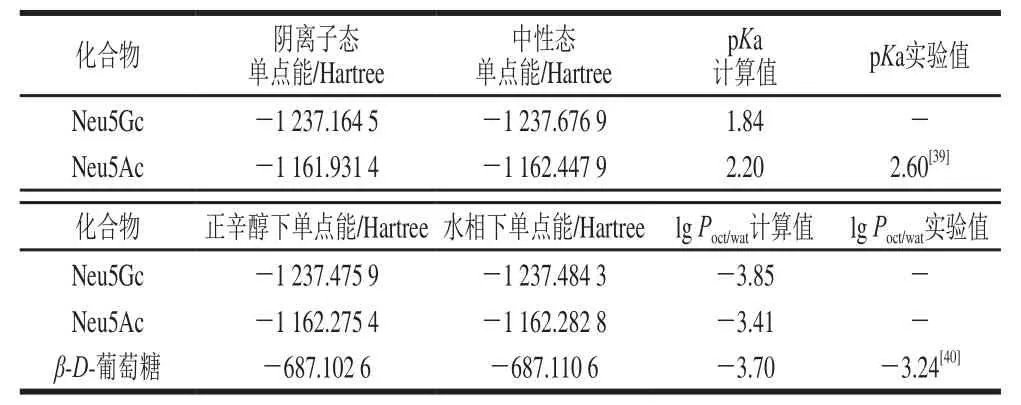
表4 Neu5Gc的理论pKa、lg P值Table4 Theoretical pKa and lg P values of Neu5Gc
2.2.2 Neu5Gc不同部位脱氢解离能比较
键解离能值越小表示化学键越容易断裂或形成,反应越容易发生。抗氧化剂清除自由基作用机制之一是氢原子转移机制。醇类如甲醇、正丁醇常作为有效的自由基淬灭剂,Neu5Gc含多个醇羟基,不同部位的脱氢活性因原子电荷分布、溶剂介电常数等不同会有所差异。结合食品加工溶剂环境,选取非极性溶剂和有机酸考察Neu5Gc脱氢解离能,结果如表5所示。

表5 Neu5Gc 不同羟基部位O—H解离能(M062X/def2TZVP)Table5 O—H dissociation energy for different hydroxyl sites of Neu5Gc (M062X/def2TZVP)
从表5可以看出,不同部位解离能有较大差异。除水相外,O7位脱氢解离能最小,气相条件下达到最低(427.908 9 kJ/mol),乳酸条件下次之(433.413 0 kJ/mol);苯条件下O10位具有最大的解离能(464.061 4 kJ/mol),O3位次之(458.618 2 kJ/mol);这和O7位负电荷集中、亲电性较强是一致的。解离能在气相、四氯化碳、乙酸、乳酸、甲酸条件下呈现出O3位>O2位>O7位的顺序。在水相、乙醇、苯相中则呈现出O3位>O5位>O4位>O2位的顺序。3 种有机酸中,乳酸条件下羟基脱氢解离焓均小于其余两种有机酸,表明羟基在乳酸条件下易于发生脱氢,这和乳酸条件下Neu5Gc的LUMO轨道值最大是一致的。参考几种抗氧化剂已知的最小脱氢解离能:没食子酸为329.69 kJ/mol、白藜芦醇为345.18 kJ/mol、槲皮素为369.86 kJ/mol,对比发现Neu5Gc具有一定的清除自由基能力[41]。这可能是Neu5Ac可降低脂质氢过氧化物细胞毒性的原因之一[42]。
2.3 Neu5Gc理论红外和紫外光谱的模拟

图4 Neu5Gc的理论红外光谱图Fig.4 Theoretical infrared spectrum of Neu5Gc
分子中的基团由于发生伸缩振动、变形振动等产生的红外谱图可以定性描述分子结构。在Neu5Gc优化后结构基础上进行振动分析,得到Neu5Gc的气相条件下理论红外光谱如图4所示。
通常非线性分子的振动模式为3N-6(N为原子数)[43],Neu5Gc含有117 种振动模式。通过对振动模式的势能分布分析,发现伸缩振动模式有40 种,面内弯曲振动模式有39 种,扭转振动模式有26 种,面外弯曲振动11 种。结合Multiwfn 3.5程序分析结果对光谱进行基团指认,图4从左往右依次可以看出尖峰(3 690.563 5、3 649.216 4、3 603.868 0、3 585.195 1、3 494.498 2 cm-1)和前后的几个小峰主要是Neu5Gc中羟基的O—H伸缩振动产生的。第2个尖峰(3302.4341 cm-1处)是乙酰基团的N—H收缩振动。接着是2 864.955 0 cm-1处的尖峰、2 852.951 0 cm-1处的小峰,这分别是环上和丙醇支链C—H的伸缩振动。1 800.600 2、1 659.219 7、1 524.508 2 cm-1这3 个尖峰则分别是羧基上的O=C键、乙酰基上的O=C键的收缩振动和N—H键的摆动。1 376.458 8、1 284.428 1 cm-1处的振动为亚甲基的摆动,1 247.082 4、1 167.055 7 cm-1处为C—H的摆动。1 116.372 1、1 079.026 3 cm-1处为环上羟基的摆动和C=O键的收缩振动。988.329 4 cm-1附近的连续小峰为C—H摆动。664.221 4、564.188 1、481.493、404.134 7、358.786 3、293.431 1、252.084 0、225.408 5 cm-1处的几个较小尖峰是羟基摆动。最后的连续弱峰是环上取代支链的弯曲振动[44]。
分子的紫外吸收光谱是分子中成键轨道的α、π电子和非键轨道的n电子吸收紫外光能量跃迁到反键轨道α*、π*、n*上的结果。Neu5Gc气相和不同溶剂环境下的理论紫外光谱结果如图5所示,不同溶剂下的最大吸收波长和摩尔吸光系数如表6所示。
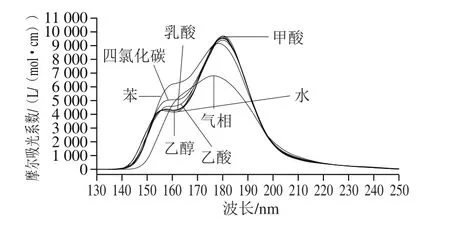
图5 Neu5Gc理论紫外光谱图Fig.5 Theoretical UV spectra of Neu5Gc
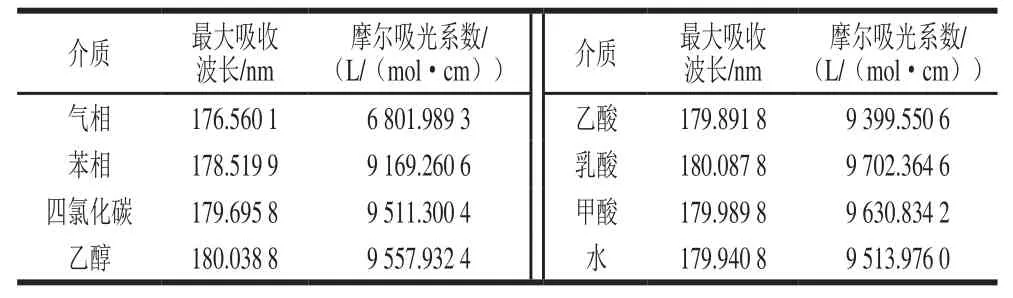
表6 Neu5Gc不同溶剂下最大吸收波长(理论)Table6 Maximum absorption wavelengths of Neu5Gc in different solvent environments (in theory)
从图5和表6可以看出,Neu5Gc的紫外光谱均分布在远紫外光区。在溶剂条件下的最大吸收波长比气相条件下大,即在溶剂环境下Neu5Gc因溶剂导致电子跃迁状态改变,发生了一定程度的红移。从图5还可看出,Neu5Gc在非极性溶剂(苯、四氯化碳、乙醇)条件下稍有差异,而在极性溶剂(甲酸、乳酸、水)下差异不大。理论最大吸收波长为180.087 8 nm,已报道的不经衍生化处理的实验最大吸收波长210、205 nm[45-46]。计算结果和实验结果的差异主要可能是由于液相紫外检测所采用的溶剂是混合溶剂、实际过程中Neu5Gc可能以混合构象形式存在,导致理论最大吸收波长产生蓝移。
3 结 论
利用量子化学理论计算对Neu5Gc进行了分子电子结构参数分析,解释和预测了其在不同溶剂下的反应活性。根据理论计算结果可知,在食品加工过程中,极性脂溶性溶剂的加工环境对Neu5Gc的反应活性影响不大,而有机酸的加入可以提高Neu5Gc的反应活性。分子表面静电势分布表明:O2位附近有负相区域极小值,羧基基团附近有正相区域极大值。EHOMO在气相条件下最小(-8.670 6 eV),ELUMO在乳酸条件下最大(1.331 2 eV)。综合亲电指数、化学势、电负性、软度、硬度得到Neu5Gc易于和乳酸等有机酸发生反应。简缩型福井函数表明氮原子的f-值均大于氧原子,羧基部位集中了较大的f+值。约化密度梯度函数和电子密度拓扑分析发现分子内氢键分布在O5—H30、O9—H31、O1—H34处,相应的距离为1.98、1.86、1.90 Å。对Neu5Gc原子自然电荷分布分析表明,负电荷集中在O7和O5位,氧原子在甲酸下易发生亲电反应。计算得到Neu5Gc的理论pKa值为1.84,lg Poct/wat为-3.85。羟基脱氢解离能计算表明,Neu5Gc有微弱的抗氧化性,气相条件下O7位最低:427.908 9 kJ/mol,乳酸条件下次之,433.413 0 kJ/mol。气相和四氯化碳、乙酸、乳酸、甲酸这4 种溶剂中解离能呈现出O3位>O2位>O7位的顺序。模拟的红外光谱图进行图谱指认后符合Neu5Gc主要官能团的振动模式,进行气相和溶剂条件下的紫外吸收光谱模拟,得到了理论最大吸收波长180.087 8 nm。
研究食品中存在的外源性致癌物(抗生素、农残、内分泌干扰物等)和内源性致癌物(生物毒素、生物胺、Neu5Gc等)的消解与迁移规律可以精准提高食品安全性。这些危害因子与生物分子、生物环境的分子间相互作用关乎人体健康,在加工过程与环境中的反应和活性变化关乎食品品质。量子化学与构效关系等理论计算已成功应用于食品抗氧化剂、食品功能因子、印迹聚合物、改性修饰、毒理学预测等的研究,提高了对反应的预测能力和机制的认识水平,为深入研究食品安全性和功能性提供了新的参考。
猜你喜欢
中华养生保健(2020年8期)2021-01-14
云南化工(2020年11期)2021-01-14
中成药(2018年2期)2018-05-09
新乡学院学报(2016年6期)2016-12-01
浙江大学学报(工学版)(2016年11期)2016-06-05
腹腔镜外科杂志(2016年12期)2016-06-01
当代化工研究(2016年9期)2016-03-20
恋爱婚姻家庭·养生版(2016年2期)2016-02-17
合成技术及应用(2015年2期)2016-01-10
河北科技大学学报(2015年6期)2015-03-11
