
Taxonomic and metabolic shifts in the Coorong bacterial metagenome driven by salinity and external inputs
2018-12-22 06:59KellyNEWTONThomasJEFFRIESReneeSMITHJustinSEYMOURLaurentSEURONTJamesMITCHELL
Kelly NEWTON , Thomas C. JEFFRIES , Renee J. SMITH ,Justin R. SEYMOUR , Laurent SEURONT , James G. MITCHELL
1 School of Biological Sciences, Flinders University, PO Box 2100, Bedford Park, SA 5042, Australia
2 School of Science and Health, Western Sydney University, Richmond NSW 2753, Australia
3 Flinders Centre for Innovation in Cancer, Flinders University, Bedford Park, SA 5042, Australia
4 Climate Change Cluster, University of Technology Sydney, Sydney NSW 2007, Australia
5 Centre National de la Recherche Scientifique, Laboratoire d’Océanologie et de Géosciences, CNRS UMR 8187 LOG, Wimereux,France
Abstract The Coorong estuary lies at the terminus of Australia’s largest river system, the Murray-Darling;both are strongly influenced by human activities; including farming and extensive flow modification.Metagenomic approaches were used to determine the planktonic bacterial community composition and potential metabolic function at two extremes in the Coorong, the river mouth which exhibits marine-like salinity, and the hypersaline upper-reaches of the estuary. Significant shifts in taxa and metabolic function were seen between the two sites. The river mouth exhibited an increase in abundance of Rhodobacteriaceae and Alteromonadaceae; families readily able to adapt to change in nutrient conditions; and the potentially pathogenic families Brucellaceae, Enterobacteriaceae and Vibrionaceae. Metabolisms over-represented include motility and chemotaxis, RNA metabolism and membrane transport, all of which are involved in actively searching for and obtaining nutrients. Also over-represented were metabolisms involved in population succession and stress response. An over-representation of taxa and metabolisms indicative of environmental change is reflective of anthropogenically affected riverine input. In the hypersaline upper reaches of the estuary, the halophilic family Ectothiorhodospiraceae was over-represented, as were the families Flavobacteriaceae, Cytophagaceae and Nocardioidaceae, members of which are able to survive over a wide salinity range. Metabolisms over-represented here were reflective of increased bacterial growth, characteristic of hypersaline environments, and included DNA metabolism, nucleotide and nucleoside synthesis and cell cycle. Coorong metagenomes clustered taxonomically and metabolically with other planktonic metagenomes, but remained an outlier of this group with only 71% and 84% similarity,respectively. This indicates that the Coorong exhibits a unique planktonic bacterial community that is influenced by riverine input at the river mouth and salinity in the upper-reaches.
Keyword: hypersaline; estuary; bacteria; taxonomy; metabolic potential
1 INTRODUCTION
Aquatic ecosystems, including estuaries, rivers,coastal embayments and wetlands, are under increasing pressure due to anthropogenic stressors such as climate change, salinization, land clearing,decreased riverine flow and rising sea levels (Jolly et al., 2001; Kingsford et al., 2011). Estuaries in particular exhibit strong changes in physiochemical properties and are extremely productive ecosystems harbouring diverse wildlife and dynamic microbial communities (Ruiz et al., 1998; Henriques et al.,2006; Kan et al., 2007), as such the impact of anthropogenic changes on estuaries is of great importance. Aquatic bacterial communities present in estuaries underpin major biogeochemical cycles and influence the community at higher trophic levels(Azam and Malfatti, 2007; Strom, 2008) yet their taxonomic composition and metabolic function is structured by “bottom-up” physiochemical factors,such as salinity and nutrient concentration and availability (Yokokawa et al., 2004; Lozupone and Knight, 2007). As a result, anthropogenic and natural processes resulting in changes in physiochemical dynamics greatly impact bacterial communities resulting in dramatic shifts in community structure and function which have flow on effects for aquatic environments.
The Coorong, South Australia, is an estuary, lagoon and lake system extending from the terminus of the Murray-Darling River system which exhibits large spatio-temporal shifts in salinity and nutrient concentrations along its length (Ford, 2007). The Coorong is a Ramsar listed Wetland of International Importance and yet is in unprecedented poor condition(Mudge and Moss, 2008). Previous research has demonstrated that aquatic and sediment bacterial community diversity and abundance in the Coorong is driven by salinity (Schapira et al., 2009; Jeffries et al., 2011, 2012), although the sediment community substrate is the major structuring factor (Jeffries et al.,2011), consistent with previous research indicating salinity is a selective pressure governing global bacterial distribution (Lozupone and Knight, 2007).However the identity and metabolic potential of the planktonic Coorong bacterial community has not yet been determined. As bacteria underpin major biogeochemical cycles, impact upon higher trophic levels and have flow-on effects for the entire ecosystem determining the identity and function of the community present and how anthropogenically riverine input and salinity effect the community is of upmost importance.
Recent advances in community profiling,specifically metagenomic approaches, have allowed for unprecedented characterisation of bacterial taxonomic composition and functional metabolic potential. In this context, the objectives of the present work were to determine shifts in bacterial taxonomic and metabolic diversity at two sites in the Coorong which exhibit marked difference in salinity from marine at the river mouth to hypersaline ~100 km to the south east in the upper reaches of the estuary, the working hypothesis being that the increase in salinity may result in shifts in the community towards taxa and metabolisms found at higher salinities.
2 MATERIAL AND METHOD
2.1 Study site
The Coorong is a shallow 2–3 km wide coastal estuarine and lagoon system extending 140 km South East from the terminus of the River Murray in South Australia (Lamontagne et al., 2004; Geddes, 2005). It is a reverse estuary and as such exhibits increasing salinity from marine at the River Mouth to hypersaline in the upper reaches of the southern lagoon. The large salinity gradient present is a result of interactions between sea level, tides, wind, rainfall, evaporation and riverine input (Webster, 2005; Ford, 2007).Temporal and spatial alterations in salinity and nutrient loading drive shifts in the abundance and flow-cytometric structure of the bacterial communities present (Ford, 2007; Schapira et al., 2009).
2.2 Sampling strategy
Samples were collected from 2 sites exhibiting the extremes in salinity found in this system; marine (Site 1; 37 Practical Salinity Scale, PSS) at the river mouth and hypersaline (Site 2; 106 PSS) near Salt Creek, in November 2009 ( Supplementary Information S1). At each site salinity (Practical Salinity Scale),temperature (°C), pH, dissolved oxygen (mg/L) and turbidity (Nephelometric Turbidity Units, NTU) were recorded using a 90FL-T multiparameter probe (TPS Ltd.). Triplicate 60 mL samples to determine chlorophylla(Chla) concentration were collected and analysed as described by Schapira et al. (2009).Triplicate 120 mL samples were collected and analysed to determine total suspended matter (TSM),particulate inorganic matter (PIM) and particulate organic matter (POM) concentration as described by Barillé-Boyer et al. (2003). Analysis of triplicate 100 mL samples for inorganic nutrient concentration was conducted using an LF 2400 photometer(Aquaspex®) and standard colorimetric methods for determining concentrations of ammonium(Indophenol Blue), Nitrite (Naphtylethylene diamine),nitrate (Naphtylethylene diamine after zinc reduction),phosphate (Ascorbic acid reduction) and silica(Heteropolyblue) as described by Schapira et al.(2009).
2.3 Microbial enumeration
Triplicate 1 mL samples for bacterial enumeration were fixed in 0.5% final concentration gluteraldehyde and stored as described by (Brussaard, 2004). Prior to analysis samples were thawed in 50°C water, diluted in 0.2 μm filtered and autoclaved TE buffer (10 mmol/L Tris-HCl, 1 mmol/L disodium EDTA, pH 8) (1:10 dilution Site 1; and 1:100 Site 2), stained with SYBR-I Green solution (Molecular Probes, Eugene, Oregon;1:20 000 dilution) and incubated at 80°C in the dark for 10 min (Brussaard, 2004; Seymour et al., 2007).Samples were then processed using a FACSCanto flow cytometer (Becton-Dickson) with 1 μm fluorescent beads (Molecular Probes, Eugene,Oregon) added to each sample at a final concentration of 105beads/mL to determine bacterial and Virus-Like Particle (VLPs) abundance (Gasol and del Giorgio 2000). Bacteria and viruses were identified and enumerated using WinMDI 2.9 (© Joseph Trotter)according to variations in green fluorescence and side scatter (Marie et al., 1999a, b; Brussaard, 2004).
2.4 Sample collection, microbial community DNA preparation and metagenomic sequencing
Microbial community DNA was extracted from 20 L of 5 μm gravity filtered and 100 kDa tangential flow filtration treated (MilliporeTM) sub-surface(approximately 30 cm deep) water using a bead beating and chemical lysis extraction procedure(UltraClean®Water DNA Isolation Kit; MO BIO laboratories, Inc.) then further concentrated using ethanol precipitation. DNA was amplified using the strand-displacement Φ29 DNA polymerase(Genomiphi V2 kit; GE Healthcare Life Sciences,Inc.) and purified using a DNeasy blood and tissue kit(Qiagen) as described by Thurber et al. (2009). DNA quality and concentration was determined by agarose gel electrophoresis and spectrophotometry and approximately 5 μg of high molecular weight DNA from each site was then sequenced by the Australian Genome Research Facility (AGRF), St. Lucia,Queensland. Sequencing was conducted on a GSFLX pyrosequencing platform using Titanium series reagents (Roche).
While multiple displacement amplification (MDA)has been shown to introduce GC bias, form multiple replication forks and chimeric sequences, and can skew the ratio of different community members(Lasken and Stockwell, 2007; Yilmaz et al., 2010),the biases introduced are minimal, with an error rate of 10-6(Esteban et al., 1993; Paez et al., 2004), and as the main focus of this study was to compare differences between Coorong metagenomes therefore any bias is applied equally (Edwards et al., 2006).
2.5 Bioinformatics and statistical analysis
Unassembled DNA sequences were annotated using the MetaGenomics Rapid Annotation using Subsystem Technology (MG-RAST) pipeline version 2 (Meyer et al., 2008). BLASTX E-value cut-off was 1×10-5and minimum alignment length was 50 bp(Dinsdale et al., 2008). Taxonomic and metabolic profiles were generated using the normalised abundance of sequences matches to the SEED database (Overbeek et al., 2005). For this analysis taxonomic information was resolved to the species level and metabolism was resolved to individual subsystems, groups of genes involved in a particular metabolic process (Overbeek et al., 2005).
To determine difference in bacterial taxonomic community structure rank abundance plots were generated. At each site the abundance of each family was normalised by overall number of sequences to give a proportion. Taxa rank was plotted on thex-axis and abundance on they-axis, both log10transformed.The data that produced the best fit then had a power law trend line regressed to those data, represented by the equation

whereais they-axis intercept andbis the slope.Significant difference between the slopes were then determined using the Student’s t test (Zar, 1996). The data not included in the power law regression, the noise/rare biosphere, is generally not included in analysis (Mitchell, 2004) and a linear trend line was regressed to these data.
Significant differences in taxonomic identity and metabolic function between the Site 1 and Site 2 metagenomes were then determined by generating an abundance summary table of frequency of hits to individual taxa or subsystems for each metagenome.The abundance summary table was imported into the STastical Analysis of Metagenomic Profiles (STAMP)software package. STAMP normalises the data to remove bias in sequencing effort and read length by dividing by the total number of hits (Parks and Beiko,2010). Taxonomy was resolved to family level and metabolism was resolved to the second level of the MG-RAST metabolic hierarchy. Fisher’s Exact test(Fisher, 1958) was used to determine correctedP-values (q-values), confidence intervals were calculated using the Newcombe-Wilson method(Newcombe 1998) and the false discovery rate was corrected for using the Benjamini-Hochberg FDR approach (Benjamini and Hochberg, 1995). To ensure only the most significant differences are reported for taxonomy the data were filtered to aq-value cut-offvalue of <1×10-15.

Table 1 GPS position, sequencing data, environmental conditions, bacterial and VLP abundance(mean±standard error), and particulate variables(mean±standard error) at each site
Similarity between our metagenomes and 26 publically available metagenomes on the MG-RAST database was determined to organism and metabolic subsystem level 3 by first generating a heatmap of the frequency of MG-RAST hits to individual genomes or subsystems for each metagenome. This was normalised by dividing by the total number of hits to remove any potential bias created by sequencing effort or difference in read length. Hits were identified using an E-value cut-off of 1×10-3to allow for difference in read length and sequencing effort. Statistical analysis was then determined using square root transformed frequency data using Primer 6 for Windows (Version 6.1.6,Primer-E Ltd., Plymouth) (Clarke and Gorley, 2006).Bray-Curtis similarity relationships between our metagenomes and the 26 publically available metagenomes were analysed using hierarchical agglomerative clustering (CLUSTER) (Clarke, 1993)and a dendogram obtained.
3 RESULT
3.1 Coorong environmental properties
Sites were characterised by a strong difference in salinity from marine conditions of Site 1 at the Murray Mouth to hypersaline conditions of Site 2 at Salt Creek (Table 1). Microbial abundances were greatest at Site 2, heterotrophic bacterial abundance increased by 12 fold, VLPs increased by 10.6 fold and Chlaincreased by 12 fold (Table 1). TSM, PIM and POM were all increased at Site 2 while inorganic nutrient concentrations were low at both Site 1 and Site 2(Table 1).
3.2 Bacterial metagenomes
Metagenome libraries were obtained from Site 1 and Site 2 in the Coorong. These libraries respectively yielded 70 353 and 47 910 reads with average read lengths of 324 and 323 bases, respectively. Both metagenomic libraries were dominated by Eubacteria with 94.8% and 92.6% hits to the SEED database,respectively (Table 1). Other domains represented in lower abundance were; Archaea accounting for 0.06%and 0.39%, Eukarya for 0.26% and 0.81% and viruses for 0.03% and 0.05% of hits respectively. Within the Site 1 and Site 2 metagenomic libraries, a total of 4.75% and 6.15% could not be assigned to any known domain.
The King s daughter shuddered11, but the King said, I have taken an oath to give you to the very first beggar-man, and I will keep it. All she could say was in vain; the priest was brought, and she had to let herself be wedded13 to the fiddler on the spot. When that was done the King said, Now it is not proper for you, a beggar-woman, to stay any longer in my palace, you may just go away with your husband. 13
3.3 Shift in taxonomic profile with salinity
At Site 1, Proteobacteria dominated, (84.4%) with a lesser contribution from Bacteroidetes (11.6%). Of the Proteobacteria, Alphaproteobacteria and Gammaproteobacteria contributed to 50.3% and 44.5% of sequences, respectively, while the Bacteroidetes were dominated by Flavobacteria, with 76.9% of sequences. At Site 2 Actinobacteria dominated (37.1%), with lesser contributions from Bacteroidetes (30.2%) and Proteobacteria (25.9%). Of the Actinobacteria, Actinomycetales represented 92.0% of sequences, Proteobacteria were dominated by Alphaproteobacteria and Gammaproteobacteria contributing 26.8% and 52.8% of sequences respectively. Bacteroidetes were dominated by Flavobacteria, (70.3%). Comparing within the Proteobacteria at each site, the orders Rhizobiales and Rhodobacterales dominated the Alphaproteobacteria at both sites. Site 1 had 16.8% and 72.8% of hits respectively while Site 2 exhibited 27.4% and 58.7%of hits respectively. The Gammaproteobacteria were dominated by the Alteromonadales (36.3%),Enterobacteriales (24.9%) and Vibrionales (15.7%) at Site 1 and the Alteromonadales (16.0%), Chromatiales(16.8%), Pseudomonadales (14.9%) and the Thiotrichales (14.2%) at Site 2. Analysis of the slope of the power law fits to rank abundance of bacterial families revealed a significant change in bacterial community structure between Site 1 and Site 2 (Fig.1)(P<0.001). The slope of the trend line at Site 1 is steeper than that at Site 2, indicating that there are a few dominant taxa at Site 1 but overall richness and diversity are greatest at Site 2 (Magurran, 2004).
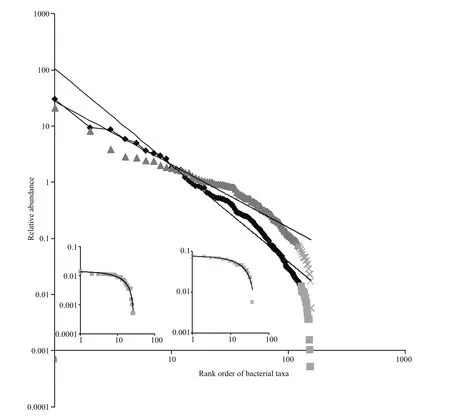
Fig.1 Rank abundance curves of bacterial community family structure showing power law trend lines at Site 1 (black diamond; y=104.8 x–1.7, R 2=0.96) and Site 2 (grey triangle; y=27.1 x–1.1, R 2=0.94); inserts show the rare biosphere with linear trend-lines, at Site 1 (grey square; y=-0.001 x+0.014, R 2=0.97) and Site 2 (grey cross; y=-0.002 x+0.077,R 2=0.97)
Fishers exact test was used to investigate significant shifts in abundance of specific taxa between the bacterial communities present at Site 1 and Site 2. At Site 1 there was an over-representation of Proteobacteria (Fig.2a), specifically Alphaproteobacteria and Gammaproteobacteria (data not shown). The alphaproteobacterial family Rhodobacteraceae and the gammaproteobacterial families Enterobacteriaceae and Vibrionaceae were over-represented by 24%, 8%, and 4.5% respectively(Fig.2b). Interestingly, there were numerous other families marginally over-represented by 2.3%±0.3%including the alphaproteobacterial family Brucellaceae and the gammaproteobacterial families Pseudoalteromonadaceae and Alteromonadaceae.
At Site 2 there was an over-representation of Bacteroidetes and Actinobacteria (Fig.2a); specifically the families Flavobacteriaceae and Microbacteriaceae were over-represented by 13 and 8% respectively(Fig.2b). Again there was a small over-representation of other families of 1.7%±0.08%, including the Bacteroidetes families Rhodothermaceae and Cytophagaceae and the Actinobacteria families Nocardioidaceae and Micrococcaceae and an unclassified family (Fig.2b). Additionally, while the class Gammaproteobacteria was not over-represented at Site 2, the gammaproteobacterial family Ectothiorhodospiraceae was.
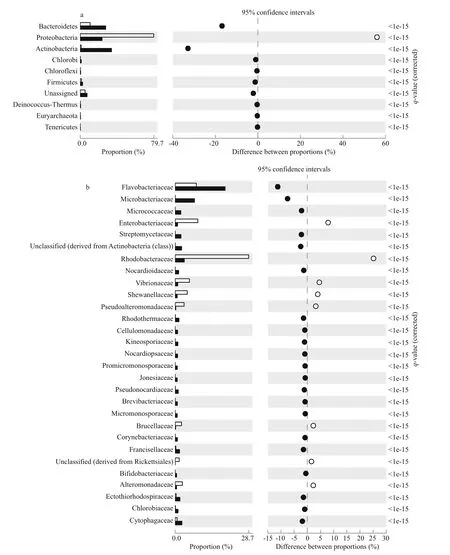
Fig.2 Phyla (a) and families (b), significantly different in abundance between Site 1 and Site 2

Fig.3 Level 1 sub-system (a) and level 2 sub-system (b) metabolic processes significantly different between Site 1 and Site 2
3.4 Shifts in metabolic potential with salinity

Fig.4 Comparison of taxonomic profiles derived from Site 1, Site 2 and 26 selected metagenomes publically available on the MG-RASTdatabase
Significant shifts in the metabolic potential of each bacterial community were further investigated using STAMP to the first, second and third levels of MGRAST metabolic hierarchy (Fig.3a, b). Genes involved in cellular response to stress, specifically oxidative stress, were over-represented at Site 1 as were processes involved in RNA processing and modification. Sequences responsible for searching,obtaining and utilisation of nutrients were also overrepresented. These included the processes of metabolism of a variety of compounds including aromatic compounds, cofactors, vitamins, prosthetic groups and pigments, di- and oligosaccharides,organic acids, fatty acids, nitrogen and sulfur metabolism. Motility, chemotaxis and membrane transport, specifically Ton and Tol transport systems,were also over-represented; however, the difference between proportions was approximately 0.25%(Fig.3a, b).
At Site 2 sequences related to increased bacterial growth and reproduction including the core metabolic functions of DNA metabolism; nucleoside and nucleotide synthesis; cell wall and capsule; protein metabolism; amino acid biosynthesis; carbohydrate metabolism; and clustering based subsystems involved in the biosynthesis of the cell wall components galactoglycans and lipopolysacharides were over-represented (Fig.3a, b). Cell division,cycle, signaling and regulation were only overrepresented by 0.35% and 0.2%. Sequences for bacterial resistance to virulence, disease and defence,specifically resistance toxic compounds were also over-represented, while sequences for iron acquisition and metabolism; photosynthesis; and dormancy and sporulation genes were also over-represented;however, the difference between proportions was again reduced (Fig.3a, b).
3.5 Comparison of planktonic Coorong taxonomic and metabolic profiles to profiles from other habitats
The taxonomic and metabolic potential of our metagenomes were compared to 26 publically available metagenomes form a variety of aquatic habitats, including fresh, marine and hypersaline waters and Coorong sediment (Table 2). Planktonic Coorong Water metagenomes were obtained from sites with vast differences in salinity, biomass and inorganic nutrient content (Table 1) and as such shared only 71% and 84% similarity to each other taxonomically and metabolically (Figs.4 and 5).Taxonomically and metabolically, the Coorong Water metagenomes form a discrete cluster with metagenomes from other marine, freshwater and estuarine environments with >65% and 84%,similarity respectively (Figs.4 and 5). When compared to metagenomes from solar salterns of varying salinity, Coorong sediment and anthropogenically affected environments clustered separately. Within the cluster that includes planktonic Coorong metagenomes, taxonomically the Site 2 metagenome showed greatest similarity to the other marine,freshwater and estuarine metagenomes (Table 2) than the Site 1 metagenome (Fig.4), while the opposite was observed for metabolism (Fig.5). Metagenomes from salterns with similar salinities to our samples, an anthropogenically affected coral reef, an aquiculture pond and samples obtained from Coorong sediment(Table 2) do not cluster with planktonic Coorong metagenomes either taxonomically or metabolically(Figs.4 and 5).

Table 2 Summary of publically available metagenomes used in this study
4 DISCUSSION

Fig.5 Comparison of metabolic profiles derived from Site 1, Site 2 and 26 selected metagenomes publicly available on the MG-RAST database
The Coorong is a dynamic ecosystem that exhibits large increases in physiochemical parameters,microbial abundance and productivity from the river mouth to the upper reaches (Ford, 2007) (Table 1).Both the River Murray and the Coorong are currently threatened by anthropogenic and climate influences(Lester and Fairweather, 2009; Kingsford et al.,2011). In the lower reaches of the river water quality is poor with irrigation return waters from dairy farms likely to be responsible for increased nutrient loads and faecal coliform abundance (Cugley et al., 2002).Inflows into the river have been greatly decreased by extended drought and water diversion resulting in the need for constant dredging from 2002 (Kingsford et al., 2011) until December 2010. In the upper-reaches of the Coorong hypersalinity is caused by reduced inflow, seawater exchange, reduced rainfall, and barrages separating the Coorong from the adjacent freshwater lakes (Webster, 2005; Ford, 2007).
4.1 Planktonic Coorong taxa shift with salinity
Salinity drives bacterial community diversity(Bouvier and del Giorgio, 2002; Wu et al., 2006;Lozupone and Knight, 2007; Tamames et al., 2010)and governs global bacterial distributions (Lozupone and Knight, 2007). Here, we show that saline and freshwater inputs play a role in shaping the bacterial community and its functional potential at two extremes of salinity in a coastal lagoon/estuary. A shift in the dominant taxa was observed between Site 1 and Site 2 confirming previous observations in the Coorong describing greater diversity in planktonic bacterial flow cytometric signatures with increased salinity (Schapira et al., 2009). Analysis of the rank abundance of bacterial families at Site 1 and Site 2 suggests that Site 1 is dominated by a few highly abundant taxa while Site 2 has increased bacterial richness and greater diversity. This is reflected in the STAMP analysis which shows a greater number of families over-represented marginally at Site 2 while only a few families are greatly over-represented at Site 1 (Fig.2b). Species diversity declines in high saline environments (Pedrós-Alió et al., 2000;Benlloch et al., 2002), the increased species diversity observed at Site 2 indicates that, although Site 2 is approximately 3 times more saline that Site 1, salinity is not high enough to reduce species diversity resulting in a unique highly exploitable environmental niche which exhibits a diverse bacterial community.
Coarse grain analysis highlights that the phyla Proteobacteria and Bacteroidetes were highly abundant at both Site 1 and Site 2 however there were statistically significant shifts in abundance of specific families which drove dissimilarity between the metagenomes (Fig.2a, b). We highlight that generally it is only over-representation of a few families that drives over-representation of an entire phyla (Fig.2a,b). While many marine bacteria phyla are ubiquitous,some families are niche specialists (Reen et al., 2006)and as such, finer levels of taxonomic resolution should be investigated when examining taxonomic shifts in planktonic environments.
Several Alphaproteobactera and Gammaproteobacteria families were over-represented at Site 1 (Fig.2b), these classes are ubiquitous in marine environments and contain many of the most abundant marine species (Biers et al., 2009; Brindefalk et al.,2011). Consequently many families over-represented at Site 1 are ubiquitous in the marine environment and have been found in other marine metagenomic studies(Jones and Betaieb, 1986; Ivanova et al., 2004;DeLong et al., 2006; Reen et al., 2006; Allers et al.,2007; Biers et al., 2009; Brindefalk et al., 2011). A strong riverine influence was observed at Site 1 where the Rhodobacteriaceae and Alteromonadaceae were over-represented. These families are known to respond quickly to allochthonous nutrient input and their over-representation is likely an indication of disturbance events in coastal surface waters (Allers et al., 2007). Members of these families plus the Pseudoalteromonadaeceae respond to increased nutrients regardless of concentration (Nogales et al.,2011). Additionally Brucellaceae, usually associated with the soil microbiota (Garrity et al., 2005) and several potentially pathogenic families, Brucellaceae,Enterobacteriaceae and Vibrionaceae (Jones and Betaieb, 1986; Garrity et al., 2005; Reen et al., 2006)previously associated with anthropogenic stress(Dinsdale et al., 2008; Nogales et al., 2011) were also over-represented . This indicates that the community at Site 1 is heavily influenced by increase in faecal coliforms, nutrient loads and other pollutants present in terrestrial runoff (Cottrell et al., 2005; Hill et al.,2005) in the lower reaches of the River Murray(Cugley et al., 2002) and riverine input has a significant impact upon the community composition.
Increased salinity at Site 2 resulted in the overrepresentation of halophilic families including: the mainly halophilic Ectothiorhodospiraceae (Imhoffand Süling, 1996; Tourova et al., 2007);Flavobacteriaceae, Cytophagaceae and Nocardioidaceae which include known halophillic members (Dobson et al., 1993; Lee et al., 2008); and the Rhodothermaceae whose members are taxonomically similar to the hypersaline genera Salinibacter (Pǎsić et al., 2009) (Fig.2b). The Bacteroidetes were also over-represented at Site 2.This phylum is diverse and widespread in marine environments (Rusch et al., 2007; Stevens et al.,2007; Tamames et al., 2010). It is also present in hypersaline salterns (Benlloch et al., 2002) and its abundance increases in productive systems (Alonso-Sáez et al., 2007) with Flavobacteriaceae as major contributors to mineralisation of primary-produced organic matter (Bowman and Nichols, 2005). As a result over-representation is a likely due to increased phytoplankton biomass at Site 2 (Table 1).
Actinobacteria are a diverse group of degraders common in soil and marine sediment (Magarvey et al., 2004; Bull et al., 2005; Stach and Bull, 2005) and were originally thought to translocate to marine environments via terrestrial run off (Bull et al., 2005).Over-representation at Site 2 of the families Microbacteriaceae, Micrococcaceae and unclassified Actinobacteria confirms recent evidence that Actinobacteria are resident members of aquatic communities (Han et al., 2003; Cottrell et al., 2005;Rusch et al., 2007; Stevens et al., 2007) and hypersaline environments (Ghai et al., 2011).
4.2 Metabolic function in the Coorong
Metabolisms over-represented at Site 1 indicate the river mouth is highly competitive environment and bacteria must exhibit the ability to utilise many metabolic pathways in order to exploit changing nutrient conditions. Metabolic processes overrepresented at Site 1 related to the metabolism and degradation of a variety of nutrient sources including sugars, fatty-acids, urea and aromatic compounds(Fig.3a, b). Processes relating to prosthetic groups were also over-represented including, tetrapyrrole derivatives responsible for chlorophyll, niacin,cobalamin and coenzyme B12 synthesis (Fig.3a, b).Prosthetic groups perform a wide range of functions(Raux et al., 2000), B12 in particular is postulated to be associated in population succession (Starr et al.,1957) supporting evidence for the over-representation of taxa that respond quickly to and are able to exploit changes in nutrient concentrations seen at Site 1(Fig.2a, b).
Increased nutrient acquisition by highly responsive taxa at Site 1 results in the creation of reactive oxygen species through the processes of nutrient oxidation,metabolism and cellular respiration (Cabiscol et al.,2000) and is likely to be responsible for the overrepresentation of cellular responses to oxidative stress, specifically glutathione regulation which is a major component of the oxidative stress response(Klatt and Lamas, 2000). Polyhydroxybutyrate metabolism was also over-represented and has been associated with increased bacterial oxidative stress tolerance environments subject to change (Ayub et al., 2004). To compete successfully for nutrient resources efficient nutrient uptake is required, this is achieved through over-representation of tRNA modification processes (Fig.2a, b) in bacteria and archaea, these processes increase both gene regulation,expression and metabolic control resulting in more efficient nutrients uptake (Persson, 1993). Membrane transport processes including Ton and Tol transport systems over-represented also indicate active bacterial community nutrient uptake. Behavioural adaptations indicative of increased nutrient competition were also over-represented including flagellar production and chemotaxis related metabolisms (Fig.3a, b). Flagella and chemotaxis assist bacteria to successfully acquire nutrients, avoid predators and other potentially detrimental situations (Blackburn et al., 1998;Kiørboe and Jackson, 2001; Pernthaler, 2005).
Hypersaline aquatic environments are characterised by an increase in microbial abundance (Guixa-Boixareu et al., 1996) consequently the majority of coarse and fine grain metabolic processes overrepresented at Site 2 were related to bacterial reproduction, growth and cellular activity (Fig.2a, b).An increase in Actinobacteria may also be related to over-representation of genes related to dormancy and sporulation and resistance to toxic compounds(specifically cobalt zinc and cadmium) as marine Actinobacteria are known to form spores (Mincer et al., 2005) and halophilic heavy metal tolerant Actinobacteria able to adapt to extreme conditions have been described previously (Nieto et al., 1989;Sarikhan et al., 2011). Metabolic processes related to increased viral abundance including phage replication and phage plasmid machinery were also overrepresented, consistent with increased viral abundance(Table 1, Fig.3a). Interestingly, it is important to note that while some bacterial families with halophilic members were over-represented at Site 2 (Fig.2a, b)generalised metabolic processes related to osmotic stress were not over-represented and only a small over-representation in the synthesis of the osmoprotectant betaine was observed (Roberts,2005). This pathway was also over-represented in hypersaline Coorong sediment in an earlier study(Jeffries et al., 2012) and indicates that as bacteria move into an environment they readily adapt to the prevalent conditions (Herlemann et al., 2011).
4.3 Taxonomic and metabolic similarity to other metagenomes
Although planktonic Coorong metagenomes were sourced from two very different salinities in the system, they were taxonomically and metabolically most similar to metagenomes from other marine,freshwater and estuarine environments. Taxonomically the Site 2 metagenome was most similar to this marine/freshwater/estuarine cluster while metabolically the Site 1 metagenome was most similar (Figs.4 and 5). Both metagenomes consistently had deeper branching nodes, branched out separately and generally were the least similar to other metagenomes in the cluster. This indicates that while the planktonic Coorong community is similar to marine, freshwater and estuarine communities it is a distinct ecosystem, with dissimilar communities,created as a result of biogeochemical properties and human activity and as such selects for a divergent bacterial community structure which exhibits functions specific to survival in this system.
Notably metagenomes from steady state,unconnected, sediment or anthropogenically influenced environments consistently clustered together, separate from the marine, freshwater and estuarine clade containing planktonic Coorong metagenomes (Table 2). The exception being aquifer metagenomes which clustered taxonomically with this clade yet were most dissimilar (Figs.4 and 5).That planktonic and sediment Coorong metagenomes are dissimilar and cluster separately is expected given that substrate type is important in structuring microbial communities (Lozupone and Knight, 2007; Jeffries et al., 2011) and indicates little transference of taxa between water and sediment and different metabolic potential. Lack of environmental connectivity also appears to be an important influence on microbial communities. Lower metabolic rates in aquifers are thought to be a result of reduced connectivity(Chapelle and Lovley, 1990), here aquifer and other unconnected metagenomes show least metabolic similarity to the marine/freshwater/estuarine metagenomes. Further evidence for this is seen in the dissimilarity of saltern and planktonic Coorong metagenomes which while similar in salinity, have markedly different environmental connectivity. The Coorong receives rainfall, riverine and marine input(Geddes, 2005) while salterns are steady state systems(Guixa-Boixareu et al., 1996) controlled within strict biochemical limits (Rodriguez-Brito et al., 2010)whose communities decrease in diversity with increased salinity driving metagenomic dissimilarity(Benlloch et al., 2002). Anthropogenic influences have previously been shown to be important in structuring bacterial communities and their functions(Dinsdale et al., 2008; Nogales et al., 2011) and in conjunction with extremes in biogeochemical conditions in the Coorong likely explain why planktonic Coorong metagenomes consistently show the least similarity to other marine/freshwater/estuarine environments and exhibit a community with a strong legacy of input source.5 CONCLUSION
Here we show the Coorong exhibits a unique bacterial consortium which is most similar taxonomically and metabolically to other marine communities despite the large differences in physiochemical properties between the two sampling sites.Significant differences in taxonomic composition are driven by anthropogenically effected riverine input and salinity, while metabolic differences are reflective of a highly adaptive community able to exploit varied nutrient sources and salinity driven increased biomass in the upper reaches of the estuary.
6 DATA AVABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
7 ACKNOWLEDGEMENT
The authors gratefully acknowledge B. Roudnew and T. Lavery for informative discussions, A. Burley and E. Alvino for editorial assistance. We sincerely thank A. Fitch from the School of Biological Sciences,Flinders University for technical support and guidance during laboratory work and S. Bailey and E. Ng from the Flow Cytometry Unit of the Flinders Medical Centre for providing technical support during flow cytometry sessions. We would like to especially thank the two anonymous reviewers whose comments and insights improved this manuscript. We also thank the Department of Environment and Natural Resources(DENR) for allowing us access to the Coorong National Park (permit number G25583-2). This research was supported by the Australian Research Council and by Flinders University. K. Newton was in recipient of a Flinders University Research Scholarship (FURS) at the time the research was undertaken.
Electronic supplementary material
Supplementary material (Supplementary Information S1) is available in the online version of this article at https://doi.org/10.1007/s00343-018-7387-z.

 Journal of Oceanology and Limnology2018年6期
Journal of Oceanology and Limnology2018年6期
- Journal of Oceanology and Limnology的其它文章
- Neuroanatomy and morphological diversity of brain cells from adult crayfish Cherax quadricarinatus*
- On the influence of season and salinity on the phenology of invertebrates in Australian saline lakes, with special reference to those of the Paroo in the semiarid inland
- Man-made plutonium radioisotopes in the salt lakes of the Crimean peninsula
- Cladophora mats in a Crimean hypersaline lake: structure,dynamics, and inhabiting animals
- Antimony speciation at the sediment-water interface of the Poyang Lake: response to seasonal variation*
- Seasonal variations of phosphorus species in the Tuohe River,China. Part I. Sediments*