
DNA的合成、组装及转移技术
2018-11-22 01:32:48卢俊南罗周卿姜双英杨焕明元英进戴俊彪
中国科学院院刊 2018年11期
卢俊南 罗周卿 姜双英 沈 玥 吴 毅 杨焕明 元英进 戴俊彪*
1 中国科学院深圳先进技术研究院 合成生物学研究所 合成基因组学研究中心 深圳市合成基因组学重点实验室深圳 518055
2 华大基因(深圳) 深圳 518083
3 天津大学 化学工程与技术学院 教育部系统生物工程重点实验室 天津 300072
随着测序技术的发展及测序成本的降低,越来越多物种的基因组被测序,人类对生物基因组的测序“读取”取得了空前的突破。而随着 DNA 合成技术及基因编辑技术的发展,人类“编写”生物基因组也逐渐成为现实。基因组的编辑,甚至全基因组的重设计与合成,一方面可以作为研究手段探索基因的功能,促进功能基因组学的发展;另一方面则可以获得新的生命体用于疾病治疗、药物生产等,服务人类。全基因组的从头合成作为当前合成生物学研究的热点之一,其涉及基因组的从头设计、构建和功能表征等,属于自下而上的生物学研究策略。新生命体系的从头设计与合成不仅需要合成组成基因组的小片段 DNA 片段,还需要通过后续的组装与拼接获取完整的合成基因组,随后还涉及合成基因组往宿主细胞的转移及功能分析。DNA 合成及基因组拼装、转移技术是合成基因组学乃至整个合成生物学领域的核心技术体系,其突破将极大地推动合成生物学的发展。
1 DNA合成技术
DNA 合成技术是合成基因组学,乃至现代分子生物学的根基。PCR(聚合酶链式反应)或者酶切手段只局限于获得自然界已有的 DNA 片段,而 DNA 的从头合成则可以通过 Oligo(寡链核苷酸)的拼接获得人工设计的特定 DNA 片段。如何提高 Oligo 合成的长度和效率、降低合成成本是后续进行大规模基因组合成的关键突破点。根据合成原理,目前 Oligo 的合成方法可分为已经成熟并商业化的化学法和正在研发中的酶促合成法。
1.1 化学法
Oligo 的化学合成研究始于 20 世纪 50 年代,Michelson 和 Todd[1]首次报道采用磷酸二酯法实现了寡聚二核苷酸的合成。到 20 世纪 80 年代,Beaucage 和 Caruthers[2]开发了基于亚磷酰胺的 DNA 合成法,也是今天 Oligo 自动化生产所采用的主要方法。该方法包括去保护、偶联、加帽(可选)及氧化 4 个步骤(图 1)。由于随着链延长所带来的化学反应效率、合成纯度以及产率的下降,目前该方法合成的 Oligo 长度一般不超过 200 个核苷酸(nt)。为了提高通量,从 20 世纪 90 年代开始发展起来的基于微阵列芯片的 DNA 合成策略,使得合成成本降低了若干个数量级[3]。然而由于芯片特有的不均一性及边缘效应等原因,相较于柱法合成,芯片法合成在长度、准确性方面均有所下降。为了增加芯片合成的精确度,2010 年 Kosuri 等[4]和 Matzas 等[5]分别采用不同的技术策略,从各异的芯片产物中挑选出正确合成的寡核苷酸原料。Kosuri 采用了多重 PCR 策略,选择性扩增目的寡核苷酸片段,结合酶促纠错等方法,可以合成长度超过 200 nt 的寡核苷酸原料,实现了更大规模和更高精度的合成[4,6];Matzas 等则是借助 454 测序仪,先通过测序挑选出序列正确的寡核苷酸产物,然后对这些产物进行大量扩增,从而使得样品的错误率降低到基本可以忽略[5,6]。
1.2 酶促从头合成法
目前亚磷酰胺法是商业化 Oligo 合成的主流方法,但其合成的长度限制在 200 nt 左右,而且合成过程中亦会大量使用有毒化学试剂[7]。由于在合成准确性及不需要使用毒性化合物等方面的潜在优势,酶促合成方法受到了越来越多的关注。酶促法从头合成寡聚核苷酸的提出至少可以追溯到 20 世纪 60 年代[7-9]。与化学法相比,酶促法的作用条件温和,对 DNA 的损伤较少,有助于准确性的提高,同时减少了副产物的产生,可实现更长 Oligo 的合成[7]。然而,酶促法的发展缓慢,至今未能实现商业化[9]。根据 Jensen 和 Davis[9]对 DNA 酶促从头合成发展的总结,末端脱氧核苷酰转移酶(TdT)介导的酶促合成法是较好的选择,但还需要更进一步优化。TdT 介导的 DNA 合成技术开发中首先需要解决单个 dNTP 添加后的终止效率及末端重新活化的问题。近期,Palluk 等[7]提出了一种解决方案,他们将 dNTP 通过可光诱导剪切的连接子与 TdT 连接,所得 dNTP-TdT 复合物可在 10—20 s 的时间完成 DNA 链的延伸,而且可以重复进行,实现特定 DNA 链的合成。该方法将为具有实际应用价值的酶促 DNA 合成法的开发打下基础。
2 合成 DNA 拼装技术
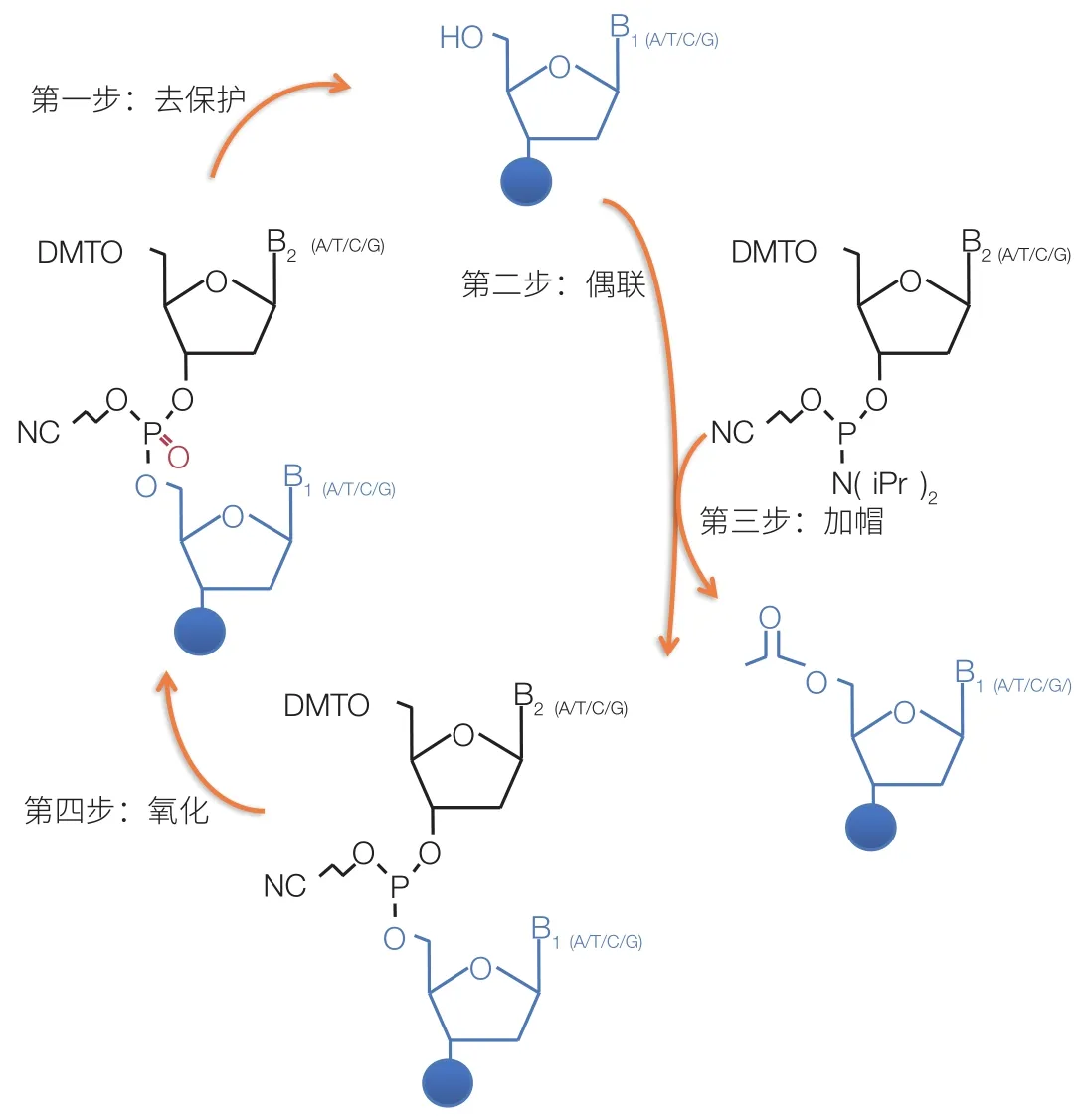
图1 固相亚磷酰胺法从头合成寡聚核苷酸链的4步反应

图2 基于不同工具酶的胞外DNA组装方法
受当前合成技术的限制,目标基因组只能以短链 Oligo 的形式得以从头合成,依赖后续的逐步拼接才能获得。根据原理区别,以下对现有的 DNA 拼接技术进行了分类总结。
2.1 胞外组装
根据组装片段的大小、序列特性、是否接受额外序列残留等,目前有众多的策略可以采用。而众多体外 DNA 组装技术的共同点在于均需要工具酶的使用,以实现 DNA 链的切割、单链黏性末端的产生、双链连接及缺口的补齐等。本文根据所用工具酶体系的不同将其归为 5 类。
(1)基于DNA聚合酶的策略。该方法首先合成有互补配对重叠区的 Oligo,利用 PCR 扩增的原理,可以对有互补重叠区的 Oligo 进行延伸连接获得小 DNA 片段,然后小 DNA 片段以重叠 PCR 的方式进行逐步连接获得目的 DNA 片段,以其作为模板,进一步 PCR 扩增可以大量获得目的片段(图 2a)。此方法(polymerase cycling assembly,PCA)无须依赖额外的 DNA 连接酶,直接从人工合成的 Oligo 开始组装,操作简单、快速。Stemmer等[10]曾用此法实现基因及质粒的一步拼装。Smith 等[11]则通过增加 Taq 连接酶的步骤用此法合成了 ФX174 噬菌体的基因组。
(2)基于同尾酶的BioBrick法和BglBrick法。同尾酶是识别不同的 DNA 序列但是能切出相同黏性末端的一类限制性内切酶,比如 XbaI 和 SpeI,但其切开的末端相互连接后不会再形成原酶切位点。利用这种“单向”的连接特性,可设计多轮连接以实现 DNA 片段组装。根据此原理设计的 BioBrick(生物积块)法[12](图 2b)可用于合成生物学中相关元件的标准化组装,但因其连接处会留下能编码终止密码子的痕迹序列,故不适用于融合蛋白的组装。为此 Anderson等[13]采用 BglII 和 BamHI 替代 XbaI 和 SpeI,其切割末端连接后产生的痕迹序列可以编码对大多数融合蛋白无影响的“甘氨酸-丝氨酸”连接肽,此策略命名为 BglBrick 法。
(3)基于 IIS型限制性内切酶的策略。BioBrick 和BglBrick 法虽然可以实现高效率的基因部件组装,但是其无法避免引入额外的痕迹序列。除了同尾酶,限制性内切酶中还存在一类 IIS 型限制性内切酶,其识别位点和切割位点不在同一个位置,因此可以根据需要放置内切酶的识别位点,以产生所需的黏性末端,用于多片段的无缝连接。2008 年,Engler 等[14]根据此原理设计了 Golden Gate 拼接法,其可在一个反应体系里面实现多片段的高效无缝连接(图 2c)。笔者实验室在 Golden Gate 拼接法的基础上,设计了 YeastFab[15]和 EcoExpress[16]两个组装系统,用于工程细胞的代谢通路优化和蛋白表达,可达 90% 以上的 DNA 拼装效率。
(4)基于多工具酶联合体系。无论是基于同尾酶还是 IIS 型限制性内切酶的连接法,由于其产生的黏性末端碱基数有限,难以支持更大片段的连接。实际上,体外的 DNA 片段连接,其核心都在于单链重叠区的产生和利用。为克服内切酶产生黏性末端长度不足的缺陷,Gibson 等[17]直接放弃限制性内切酶,采用 5′ 核酸外切酶,联合 DNA 聚合酶以及 DNA 连接酶,开发了 Gibson 组装法(图 2d)。此方法一方面可以实现无缝连接,没有痕迹序列的残留;另一方面其能连接获得的片段大小可达几百 kb(千碱基对),大大提高 DNA 体外组装所能达到的量级。
(5)DNA元件的标准化设计。为了最大化的降低对 DNA 片段的重新合成,提高对已有 DNA 片段的利用率,合成生物学的一个重要研究方向是推进合成片段(生物元件)的标准化。上述提及的 BioBrick 就是标准化的实现形式之一,是合成生物学领域最早建立的 DNA 标准化组装方法。但是 BioBrick 法在被连接的元件之间留下的痕迹不利于蛋白元件的组装。BglBrick 法就是为解决 BioBrick 的缺陷而产生的标准化策略。我国王金课题组用归位内切酶代替常规的 II 型限制性内切酶建立了 iBrick 标准[18],同时基于 CRISPR/Cpf1 技术开发了 C-Brick 拼接标准[19]。基于 IIS 型限制性内切酶的Golden Gate 标准,包括 MoClo[20]和 GoldenBraid 2.0[21],也能通过统一的方式进行元件的组装。针对 Golden Gate 等标准的不足,王金课题组建立了 MASTER 连接法[22]以实现更大片段的无缝克隆。此外,还有与 iBirck 标准类似的 HVAS 法[23],与 MASTER 类似的 GreenGate 法[24]等。不同的实验室如果都按照上述的方式对生物元件进行标准化,将能促进生物元件的流通和共享,提高复杂生命系统合成的效率。
2.2 胞内组装
尽管体外拼装的片段大小可达几百 kb,但是所得的量往往不足以进行后续实验,需要使用大肠杆菌等进行扩增。对于 Mb(百万碱基对)级别的体外组装尚未见报道,即使假设能体外组装成功,可能也无法导入到大肠杆菌内进行扩增。而枯草芽孢杆菌、酵母及工程改造的部分细菌(超表达 T4 DNA 连接酶或整合 λ Red 重组酶)等微生物本身就可以介导 DNA 的胞内重组连接[25],可直接将需要组装的 DNA 片段导入这些宿主细胞内进行组装。酵母作为常用的 DNA 重组宿主,其高效的同源重组性能早在 40 多年前就已被发掘[26-28]。1991 年,Silverman 等[29]报道酵母可以实现长达 2 Mb 的人工染色体组装。而 2010 年,Gibson 等[30]报道合成世界上第一个人造生命体,其长达 1.1 Mb 的合成基因组也是由酵母拼装获得。借助于酵母强大的同源重组能力,Sc2.0 项目(酵母基因组合成计划)[31]利用 SwAP-In 的方法实现了天然染色体的置换,获得完全合成的人工染色体[32-37]。2018 年 8 月,Shao 等[38]报道其将酿酒酵母 16 条染色体合并为 1 条长度达 11.8 Mb 的染色体,并获得有正常功能的单染色体酵母菌株。带有 1 条染色体的酵母,可作为一个新的研究平台,增进我们对染色体重组、复制和分离机制的解析,具有重要的意义。此外,该研究的结果也说明酿酒酵母对染色体长度惊人的容忍度(至少可以长达 12 Mb),这为利用酵母构建高等生物的超长染色体提供了理论依据,有利于后续 GP-write 项目(基因组编写计划)[39]的开展。
3 合成染色体转移技术
由于需要进行基因组合成的生物体本身存在生长速度慢,DNA 重组能力不足,或者转化效率低等问题,难以直接在目标细胞中进行基因组的拼装。这也是为何要使用大肠杆菌或者酵母等微生物进行拼装的原因。随着合成基因组学从低等生物向高等生物的拓展,除了更大合成染色体拼装带来的挑战,超大染色体的转移也将是一项艰巨的任务。相关报道显示,阳离子脂质和聚合物[40]、显微注射[41]、微细胞法(microcell-mediated chromosome transfer,MMCT)[42]都可以介导 Mb 级别的染色体转移。此外,电转化也是经常用于细胞系及原代细胞的转染方法,尤其能胜任对脂质体转染法等有抵抗性的细胞的转染[43]。在细菌方面,电转化可介导超过 700 kb 的 BAC 的转化[44]。聚乙二醇(PEG)介导的裸 DNA 转移法也是常用的 DNA 转染法,Gibson 等[30]成功利用此法将丝状支原体长达 1.1 Mb 的人工合成染色体移植到山羊支原体受体细胞,获得人类史上首个人工合成的生命体。然而,阳离子聚合物、PEG等基于化合物的转染方法,显微注射、电转化等物理操作,以及微细胞介导的转染方法均有其局限性。
PEG 除了可以直接介导裸 DNA 的转染,还可以通过诱导细胞融合实现间接转染,即 PEG 介导的细胞融合法。例如,PEG 可以介导酵母原生质体球与哺乳动物细胞的融合,从而将位于酵母细胞的酵母着丝粒质粒转移到受体细胞。细胞融合可以绕开受体细胞膜的阻碍,直接将载体送入胞浆,但是受体细胞核核膜依然是一个屏障,因此此法也面临效率低的问题[45]。Brown 等[45]认为,将受体细胞同步化至 M 期(有丝分裂期),此时的细胞核膜和骨架正处于重塑状态,有望提高转移的效率。实验证据表明,利用同步化到有丝分裂期的哺乳动物细胞进行膜融合转移,效率可以提高近 300 倍,且不受被转移载体大小的影响[45]。PEG 介导的细胞融合法可直接利用酵母系统进行 Mb 级别的合成染色体体内组装,因此不需要载体的分离纯化,可以避免载体受到剪切力的损伤,并且其效率受转移 DNA 大小的限制不大。基于以上优点,对此法进行改良来进一步提高其转移效率将更能满足越来越高的合成染色体移植要求。
4 野生型基因组清除技术
合成生物学目前尚未能完全从头合成一个完整的细胞结构,只能借用已有的宿主细胞。为获得只含有人工合成基因组的生命体,需要采取一定的策略将野生型染色体清除。
4.1 基于负筛选的染色体清除策略
即使在人工染色体已经植入宿主细胞并能发挥功能的情况下,对应内源染色体在自然状态下丢失的概率依旧极低,需要通过筛选才有可能获得这类细胞。Li等[46]曾报道用正负筛选联合的策略成功去除 21 三体综合征患者诱导多功能干细胞(iPSCs)中的一条 21 号染色体,使其核型恢复正常。他们把一个同时编码负筛选标记和正筛选标记的双功能融合基因敲入到 21 号染色体;通过正筛选获得融合基因整合的细胞株,并从中进一步筛选获得融合基因单拷贝的细胞株;接着在无正筛选药物的条件下培养,产生携带融合基因的染色体丢失的细胞。由于负筛选基因能将无毒的负筛药物转化成有毒的物质,进而把宿主细胞杀死,故可以通过负筛选富集获得一条 21 号染色体丢失的细胞株。通过正负筛选策略进行合成染色体转移后的内源染色体的清除,需要依赖于首先将正负筛选融合基因插入指定的内源染色体。近年来,基因编辑技术的飞速发展有望进一步提升该策略的效率。
4.2 Cre-loxP 介导的染色体清除
Cre是一种重组酶,能识别 DNA 上的 loxP 位点,并能根据两个 loxP 位点的排列方向将其之间的 DNA 片段进行删除、倒置等。此方法也可用于内源染色体的清除。通过与鼠胚胎干细胞(ESCs)融合,人成体细胞细胞核可以被重编程,获得多能性,但需要在重编程后将 ESC 来源的相关染色体清除。为达到此目的,Matsumura 等[47]设计了一个基于 Cre-loxP 重组系统的策略——CEC(chromosome elimination cassette)。CEC的中部携带一个荧光报告基因和一个抗药筛选标记,其两端分别加入一个 loxP 位点,二者相向排列。Cre 介导的姐妹染色体重组,可以产生双着丝粒染色体和无着丝粒染色体,此类异常染色体可在细胞分裂中被清除。然而,与基于正负筛选融合基因的策略类似,都需要事先对目标染色体进行相关基因的敲入操作。
4.3 CRISPR/Cas9介导的染色体清除
CRISPR/Cas9 是基于细菌 II 型 CRISPR/Cas 免疫防御系统开发的基因编辑技术[48],由于其操作简单,并能高效介导精确的基因敲除、敲入等而被广泛用于各物种的基因编辑研究。鉴于上述方法的复杂和低效性,Zuo 等[49]尝试将 CRISPR/Cas9 技术用于染色体的靶向清除。CRISPR/Cas9 系统包括两个核心部件,本身无靶向性的 Cas9 核酸内切酶以及引导 Cas9 进行靶向切割的引导 RNA(single guided RNA,sgRNA),Cas9 和 sgRNA 结合,以复合物的形式在特定位置切断双链 DNA。他们发现通过多个位点的 CRISPR/Cas9 靶向切割,细胞系、胚胎和体内组织的性染色体,以及肿瘤细胞等的常染色体能被选择性清除。这个方法可为特定染色体缺失动物模型的构建,以及相关人类遗传病的治疗提供新的策略[49],也是合成基因组学中内源染色体靶向清除的潜在技术手段。
5 结语
当前,全基因组合成已经在病毒及细菌上获得成功,首个全人工合成的真核生物——Sc2.0 也已经接近完成,对于更高等生物的全基因组合成也已经提上日程[39]。然而,由于基因组极其庞大,要实现这些基因组的合成将依赖于上述各项技术的突破以及新技术的出现。
获得自然界不存在的基因组,DNA 合成是目前唯一的手段;对于庞大基因组的合成需要更高效率、更高精度的 DNA 合成技术,同时需要进一步降低合成成本。与化学法相比,DNA 的酶促合成有着诸多优势,但是距离商业化应用尚需时日。在大基因组的设计与合成中,如果可以直接使用自然界存在的 DNA 片段,可以节省 DNA 合成的成本,但同样需要相应的技术支撑。常规长度的 DNA 片段可以通过 PCR 或者酶切等方式获取,但对于超大片段,这些策略难以胜任。CRISPR/Cas9 技术的出现使得直接从天然基因组中特异切割获取大片段 DNA 成为可能。例如,我国清华大学朱听和中国科学院微生物研究所娄春波等就联合开发了基于 CRISPR/Cas9 和 Gibson 组装的克隆策略,可获得长达 150 kb 的DNA 片段[50]。
在合成基因组拼装方面,酵母由于本身具备很强的 DNA 重组能力,是合成基因组学的重要分子操作工具,而对于未来超大基因组的合成,酵母能否胜任需要我们探讨验证。此外,合成基因组的分离提取,转移技术都会随着目的基因组的增大而面临效率减低,甚至不适用等问题,新型技术亟待开发。野生型基因组的清除是获得完整基因组合成生物体必经步骤,CRISPR/Cas9 技术有望胜任超大内源基因组的清除,但对于内源基因组不同的位点,其切割效率可能不一致,可能需要优化筛选;另外,技术本身存在的脱靶效应可能会作用于植入的合成基因组,这些问题都需要在基因组设计阶段予以考虑。
当前,我国在合成生物学领域已经取得一定的成就,尤其在 Sc2.0 计划上,我国科学家作出了重大贡献。作为新兴交叉学科,除了基本技术体系,合成生物学的发展还面临其他众多技术难题,这也是我们产出原创性成果、培养交叉型人才的机遇。
猜你喜欢
今日农业(2021年11期)2021-08-13 08:53:24
科学之谜(2019年3期)2019-03-28 10:29:44
科学之谜(2018年8期)2018-09-29 11:06:46
中国调味品(2017年2期)2017-03-20 16:18:25
创新作文(小学版)(2016年16期)2016-11-11 05:47:54
恋爱婚姻家庭·养生版(2016年9期)2016-09-07 11:25:01
现代检验医学杂志(2016年5期)2016-08-20 03:17:04
中国科技信息(2015年2期)2015-11-16 08:18:32
中央民族大学学报(自然科学版)(2015年2期)2015-06-09 08:45:16
遗传(2014年3期)2014-02-28 20:58:49
