
嗜热芽孢杆菌CHB1环糊精酶基因优化及其在毕赤酵母中的表达
2018-11-06 11:15陈龙军陈济琛林晓栩邱宏端林新坚蔡海松
食品与生物技术学报 2018年9期
陈龙军, 陈济琛, 林晓栩,, 邱宏端, 林新坚, 蔡海松*
(1.福建省农业科学院土壤肥料研究所,福建 福州 350003;2.福州大学 生物科学与工程学院,福建 福州350108)
环糊精(cyclodextrins,简称CD)作为一类重要的工农业原材料,广泛运用于食品、医药、化妆品和环 保 等 领 域[1]。 而 环 糊 酶 (cyclodextrin glycosyltransferase,简称CGTase)是生物酶法生产环糊精的重要工业用酶,它属于α-淀粉酶家族中的一类复合型多功能酶[2],能够催化环化反应、歧化反应、偶合反应和水解反应。根据现有的报道,CGTase的获取方式大致归纳为3种:从自然界中筛选野生菌株;根据基因文库进行克隆表达;对已知目的基因片段进行蛋白质工程改良。其中野生菌株筛选不仅最为直接有效,而且也是后两种方式的基础[3-4]。
尽管不断有新的CGTase得到分离鉴定,但是天然菌株稳定性差、产酶量低、分离纯化困难、不易工业化等缺陷极大限制了CGTase的工业化应用。随着近代分子生物学及基因工程技术的发展,运用生物工程手段实现CGTase基因的异源超量表达成为国内外研究的热点。其中原核表达多有报道[5-7],但真核酵母系统表达CGTase鲜见报道,目前仅见两篇文献[8-9]有相关报道,表达效果亦不甚理想。毕赤酵母作为一种高效表达系统,遗传背景清晰,成功实现了多种外源蛋白质的高效表达,探索环糊精酶在毕赤酵母中表达的可行性具有重要意义。因此作者以前期从嗜热芽孢杆菌 CHB1中分离获得的新型环糊精酶基因为基础,以毕赤酵母GS115为表达系统,通过密码子优化,比较密码子调整对环糊精酶表达效果的影响,以期实现其在毕赤酵母的异源表达,为该CGTase的实际应用提供理论与技术支持。
1 材料与方法
1.1 实验材料
菌株和质粒嗜热芽孢杆菌CHB1(含新型环糊精酶基因)、Escherichia coliDH5α:作者所在实验室保存;GS115、pPICZαA:Invitrogen 公司。
试剂与仪器限制性内切酶、Taq DNA聚合酶、DNA Mark、T4DNA 连接酶:TaKaRa 公司;DNA切胶回收试剂盒、PCR引物、博来霉素及质粒快速提取试剂盒:上海生工股份有限公司;其余试剂均为国产或进口分析纯。3-18K低温高速冷冻离心机:德国Sigma公司;PCR仪:基因有限公司;蛋白质电泳仪:美国BIO-RAD公司;Multiskan FC全自动酶标仪:美国Thermo公司。
培养基
1)地芽孢杆菌CHB1生长培养基(组分g/L):大豆蛋白胨10,牛肉浸膏5,氯化钠0.2;pH 7.0。
2)低盐 LB 培养基(组分 g/L):胰蛋白胨 10,酵母浸膏5,氯化钠5;固体培养基添加2 g/dL琼脂。
3)YPG 培养基(g/L):胰蛋白胨 20,酵母粉 10,甘油20;固体培养基添加2 g/dL琼脂。
4)BMGY培养基:蛋白胨20 g,酵母浸出物10 g,加入 500 mL水,121℃灭菌 20 min,冷至室温加入灭菌的 PBS l00 mL,10×YNB 100 mL,500×B 2 mL(过滤除菌),10×GY 100 mL,定容到 1 L;
5)BMMY培养基:蛋白胨20 g,酵母浸出物10 g,加入 500 mL水,121℃灭菌 20 min,冷却至室温加入灭菌的 PBS l00 mL,10×YNB 100 mL,500×B 2 mL(过滤除菌),甲醇 5 mL,定容到 1 L。
1.2 实验方法
基因组DNA和质粒的提取地芽孢杆菌CHB1基因组DNA的提取参照Zhou J[10]的方法进行。质粒的提取参照质粒提取试剂盒说明书的方法进行(上海生工股份有限公司)。
野生型环糊精葡萄糖基转移酶基因的克隆根据地芽孢杆菌CHB1环糊精葡萄糖基转移酶基因(CGT1基因)序列,设计上下游引物如下:
CGT-F1:5'-CTGAATTCGCTGGAAATCTTAAT AAGG-3'(下划线为限制性酶EcoRI);
CGT-R1:5'-TGGCGGCCGCGTTTTGCCAATTC ACTATAA-3'(下划线为限制性酶NotI);
以地芽孢杆菌CHB1基因组DNA为模板,采用PCR方法扩增CGT1基因,扩增条件如下:95℃预变性 5 min;94℃变性 1 min,55℃退火 1 min,72℃延伸 90 S,30个循环;72℃后延伸 10 min;4℃保存;PCR产物进行1 g/dL的琼脂糖凝胶电泳检测,通过胶回收纯化PCR产物,16℃下 与pMD18-T载体进行过夜连接,连接产物转化感受态细胞Escherichia coli DH5α,挑取阳性克隆进行菌落PCR鉴定,同时提取pMD18T-CGT1质粒送至上海生工有限公司测序验证。
环糊精酶(CGT)基因密码子优化及克隆根据CGT1的氨基酸组成,考虑毕赤酵母的碱基偏好性[11],进行密码子优化,同时在CGT1基因首末端分别引入限制性酶切位点EcoR I及Not I,将优化序列CGT2送至上海生工生物工程有限公司合成,连入pMD-18T载体,构建质粒pMD18T-CGT2。
原始基因与优化基因表达载体及重组菌构建载体 pMD18T-CGT1、pMD18T-CGT2 及 pPICZαA分别用限制性内切酶EcoR I和Not I进行双酶切,分别胶回收 CGT1、CGT2基因片段及 pPICZαA线性载体;参照TaKaRa公司T4 DNA连接酶使用说明书,将CGT1、CGT2基因片段分别与pPICZαA线性载体混合进行连接反应,连接产物转化感受态细胞DH5α,经25 μg/mL博来霉素抗性LB平板及菌落PCR鉴定,构建重组酵母表达质粒pPICZαACGT1和pPICZαA-CGT2;同时提取相应质粒送至上海生工生物工程有限公司测序验证。
分别用限制性内切酶Sac I、Pme I对重组表达载体pPICZαA-CGT1和 pPICZαA-CGT2进行线性化,胶回收后,利用电转化方法导入感受态毕赤酵母GS115中,于100 μg/mL博来霉素抗性YPG平板培养至转化子出现,对转化子进行PCR验证。
重组酵母诱导表达CGTase效果比较将筛选获得的环糊精酶重组工程菌分别接种在含有100 mL YPG培养基的500 mL摇瓶中,在200 r/min、30℃下培养24 h;以5%的接种体积分数接种于含有200 mL BMGY培养基的500 mL摇瓶中,培养至OD600为 6~8。 4℃、5 000 r/min收集酵母细胞,同时补加适量BMMY培养基重悬酵母,以后每24小时取样测环糊精酶活力,并补加1%甲醇。
环糊精酶摇瓶发酵条件优化为提高环糊精酶在毕赤酵母中的表达效率,作者对影响酵母表达的重要因素(诱导温度、pH值、诱导甲醇体积分数等)进行优化。将重组菌接种于含100 mL YPG培养基的500 mL摇瓶中。在200 r/min、30℃下培养24 h,以5%的接种体积分数接种到含有BMGY培养基的250 mL的摇瓶中,在200 r/min、30℃下培养24 h;4℃、5 000 r/min收集酵母细胞,同时补加适量BMMY培养基重悬酵母。以后每24小时补加1%的甲醇进行诱导产酶。诱导温度分别设为22、25、28、30、32 ℃;pH 设置为 4、5、5.5、6、6.5、7 共六个梯度;甲醇体积分数梯度为0.5%、1%、1.5%、2%、2.5%;诱导表达时间的优化以每24小时取样测定酶活性、OD600来确定最佳发酵时间。每组设置3个平行实验,分别测定环糊精酶活性和细胞密度OD600值。
环糊精酶活性测定测定环糊精葡萄糖基转移酶活力的方法参照甲基橙褪色法[11]并作适当改进。取50 mmol/L pH 6.0磷酸钠缓冲液配制的3 g/dL可溶性淀粉溶液0.9 mL于试管中,将试管置于60℃水浴锅内预热2 min,然后往试管内加入离心得到的发酵液上清液即粗酶液或适当稀释的纯酶液 0.1 mL,反应 10 min,加入 1.0 mL、1 mol/L 盐酸溶液终止反应,再加入0.1 mmol/L的甲基橙溶液1.0 mL,振荡混匀后于16℃保温20 min,用酶标仪于波长505 nm处测定溶液的吸光度并根据空白计算褪色程度(ΔA),最后根据α-环糊精标准曲线计算出α-环糊精的质量浓度。酶活性单位定义为:60℃、pH 6.0 下,每分钟催化产生 1 μmol α-环糊精所需的酶量为一个酶活力单位。
2 结果与分析
2.1 嗜热芽孢杆菌基因组提取及原始环糊精酶基因克隆
以嗜热芽孢杆菌CHB1的基因组DNA为模板,以CGT-F1和CGT-R1为上下游引物扩增原始环糊精酶CGT1,电泳结果见图1。扩增获得大小在2 000 bp左右的特异性DNA片段,与目的基因片段大小(2 031 bp)一致。进一步进行基因测序,结果显示扩增序列与原始环糊精酶100%一致,至此,成功获得大小和序列均正确的原始环糊精酶CGT1。将克隆获得的CGT1与pMD18T连接,通过CGT-F1/CGT-R1引物进行菌落PCR验证,结果见图2。在2 kb左右获得预期大小的DNA片段,经测序后构建获得克隆载体pMD18T-CGT1。

图1 嗜热芽孢杆菌基因组及CGTase基因PCR扩增产物的电泳分析Fig.1 Agarose gel electrophoresis of CGTgene and Gebacillius sp.CHB1 genome
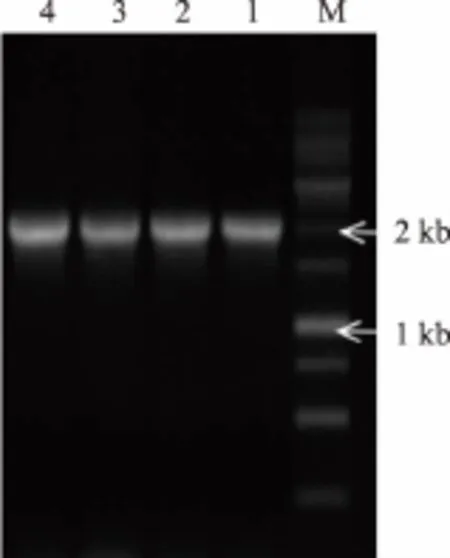
图2 克隆载体pMD18T-CGT1菌落PCR验证Fig.2 Colony PCR validation of pMD18T-CGT1
2.2 重组表达载体构建及验证
以EcoRI和NotI为双酶切体系,分别对载体pMD18T-CGT1、pMD18T-CGT2 及 pPICZαA 进行双酶切,回收 2 000 bp(CGT1)、2 000 bp(CGT2)及3 600 bp线性载体pPICZαA。将回收片段CGT1和CGT2分别与线性载体pPICZαA连接,筛选克隆子进行菌落PCR验证,电泳结果见图3。出现了预期大小的两条带;挑取2、4号克隆子提质粒送上海生工进行测序验证,由测序结果看出,原始基因CGT1和优化基因CGT2,均正确插入到pPICZαA,成功构建分泌型重组质粒 pPICZαA-CGT1和 pPICZαACGT2。
2.3 重组环糊精酶毕赤酵母工程菌构建及验证
将重组表达质粒pPICZαA-CGT1和pPICZαACGT2经相应内切酶线性化后,分别电转化毕赤酵母,提取重组酵母基因组进行PCR验证,以酵母信号肽为基础设计上游引物α-factor(5'-CAAATTCCGGCTGAAGCTGTCATC-3'), 分 别 以CGT-R1,CGT-R2为下游引物,进行原始基因与优化基因的扩增验证,电泳结果见图4。重组酵母可扩增得到大小2 100 bp,与预期大小一致,同时将PCR产物送上海生工进行测序,结果显示环糊精酶基因已成功整合入酵母染色体,并处于分泌信号肽α因子下游;至此获得两株稳定遗传的环糊精酶重组毕赤酵母工程菌,GS115/pPICZαA-CGT1和GS115/pPICZαA-CGT2。
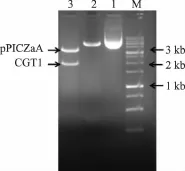
图3 重组载体pPICZαA-CGT构建Fig.3 Agarose gelelectrophoresisofpPICZαACGT1and pPICZαA-CGT2

图4 重组酵母基因组PCR鉴定结果Fig.4 PCR of recombinant Pichia pastoris
2.4 重组菌诱导产酶效果比较
以相同的诱导条件,诱导重组酵母GS115/pPICZαA-CGT1与 GS115/pPICZαA-CGT2 表达环糊精酶,比较密码子优化前后环糊精酶的活性变化。由图5产酶曲线可看出,诱导96 h后酶活力趋于稳定,经过120 h诱导后,原始环糊精酶CGT1酵母胞外表达活性达到最高,为0.37 U/mL,是张佳瑜[9]等表达的环糊精酶活性的1.6倍。而优化基因CGT2经过120 h后活性达到0.62 U/mL,是优化前CGT1活性的1.7倍,说明通过碱基偏好性的优化,将原始基因中的低频密码子优化成酵母表达系统中的高频密码子,有利于提高环糊精酶在毕赤酵母中的表达量。

图5 密码子优化前后环糊精酶工程菌表达效果比较Fig.5 Comparison of cyclodextrin enzyme activities between GS115/pPICZαA -CGT1andGS115/pPICZαA-CGT2
2.5 重组菌发酵条件优化
对碱基优化后的环糊精酶工程菌GS115/pPICZαA-CGT2进行发酵条件优化,以诱导发酵120 h为最佳诱导时间,优化包括诱导温度、pH及甲醇诱导体积分数等主要因素,结果见图6(a)。随着诱导温度的提升,环糊精酶活性逐渐升高,当诱导温度达到28℃,环糊精酶活性达到最大值0.76 U/mL,随着温度继续升高,活性逐渐下降,究其原因可能是高温加速了酵母自身蛋白酶的表达,导致环糊精酶蛋白被降解。不同pH对酵母表达外源蛋白具有重要影响,结果见图6(b)。环糊精酶胞外活性随着pH的逐渐升高,呈现先上升后下降的趋势,在pH 6.5时,活性达到最高的0.96 U/mL。甲醇作为碳源及诱导剂,其添加量对环糊精酶的表达至关重要。由图6(c)可知,随着甲醇体积分数的增加,酶活力及菌体量逐渐增加,当甲醇体积分数大于2%时,酶活力达到最大值,随后逐渐降低,可能是由于甲醇本身对酵母细胞的毒性,过量的甲醇不利于表达外源蛋白质。通过主要条件的优化,获得最佳发酵条件:pH 6.5,温度28℃,甲醇体积分数为每24小时1.5%,诱导120 h酶活力达到1.26 U/mL,是优化前的2倍。

图6 重组菌的发酵条件优化Fig.6 Optimization of fermentation conditions
3 结语
作者从嗜热脂肪芽孢杆菌CHB1中扩增获得新型CGT酶基因全序列,并实现了其在毕赤酵母中的异源表达。原始基因CGT1,在切除其自身信号肽,插入pPICZɑA表达载体的ɑ-Factor信号肽之后,转化毕赤酵母GS115,获得了基因工程菌GS115/pPICZαA-CGT1,其胞外环糊精酶活力为0.37 U/mL,高于张佳瑜[9]等表达的环糊精酶活性。碱基密码子优化后的CGT2,通过同样手段构建获得工程菌GS115/pPICZαA-CGT2,其胞外酶活力达到0.62 U/mL,是优化前CGT1活性的1.7倍;进一步对该重组菌进行发酵条件优化,确定最优条件为pH6.5、28℃、200 r/min、每 24小时补加 1.5%的甲醇诱导,120 h后其胞外酶活力达到1.26 U/mL,是优化前的2倍。
许多研究表明,外源基因密码子优化对提高外源蛋白质表达量具有显著作用,可提高几倍甚至数十倍[13-15]。本研究通过毕赤酵母密码子优化网站(http://www.kazusa.or.jp/、http://www.genscript.com等)及赵翔[11]关于毕赤酵母密码子的分析,对野生环糊精酶基因密码子在毕赤酵母中的使用频率进行分析,发现毕赤酵母低频率密码子在该基因中所占比例高达11%,经过密码子优化后低频密码子使用频率降至6%,其酵母表达活性相应提高了1.7倍,说明密码子优化对CGT的表达效率确实有一定促进作用,但表达仍不甚理想,亦低于前期大肠杆菌的表达效果。进一步通过氨基酸序列分析,发现在环糊精酶AA序列中含有多达11个潜在糖基化位点序列Asn-Xaa-Thr/Ser(其中aa代表所有氨基酸);这些位点可能导致环糊精酶在毕赤酵母内被过度糖基化,从而影响了环糊精酶的活性。下阶段考虑对酶蛋白实施去糖基化改造,考察糖基化对酶活力的具体影响;同时优化其他相关因素如信号肽、表达载体、表达宿主等,最大限度提高其在酵母中的表达效果。另外,鉴于本研究的环糊精酶基因来自芽孢杆菌,考虑通过芽孢杆菌同源表达系统实现该环糊精酶的高效表达,为环糊精酶的实际应用奠定基础。
猜你喜欢
生物学通报(2020年11期)2020-10-22
发明与创新·中学生(2019年6期)2019-06-26
中成药(2018年8期)2018-08-29
中成药(2018年7期)2018-08-04
中成药(2018年6期)2018-07-11
中成药(2018年4期)2018-04-26
中成药(2017年5期)2017-06-13
中国调味品(2017年2期)2017-03-20
现代检验医学杂志(2016年5期)2016-08-20
中国科技信息(2015年2期)2015-11-16
