
一例Crouzon综合征合并黑棘皮病患儿FGFR3基因突变分析
2018-09-06 05:30计雄飞纪超唐珊郭春燕程波
中华皮肤科杂志 2018年8期
计雄飞 纪超 唐珊 郭春燕 程波
350000福州,福建医科大学附属第一医院福建省皮肤病分院(计雄飞、纪超、唐珊、程波);福建省龙岩市第一医院皮肤科(郭春燕)
Crouzon综合征是颅缝早闭引起的颅腔狭小、眶浅和眼球突出、鹰钩鼻等颅面畸形。黑棘皮病是以皮肤色素沉着、角化过度、天鹅绒样增生为特征的皮肤角化性疾病。Crouzon综合征合并黑棘皮病(CAN)少见,由特定的成纤维细胞生长因子3(FGFR3)基因突变引起,是一种独立的疾病,有学者称之为Crouzon样皮肤骨骼综合征(Crouzono⁃dermo⁃skeletal syndrome)[1]。我们通过PCR 及 DNA 测序检测 1例CAN患儿FGFR3基因突变情况。
一、资料和方法
1.病历资料:患儿女,7岁,前额突出、突眼伴全身皮肤色素沉着7年。生后1周即发现双肘内侧色素沉着,1年后全身皮肤渐出现多处色素沉着。自幼泪管堵塞,多次疏通效果不明显。患儿父母正常,非近亲婚配,无相同疾病家族史。体检:身高135 cm,全身浅表淋巴结无肿大,心肺无异常。头颅呈方形,前额突出,上颌发育不全,下颌前凸;眼球突出,双眼间距较宽;鼻梁塌陷,上下牙咬合不良,牙列不整,参差不齐(图1A)。皮肤科检查:全身皮肤黑褐色,颈部及腋下、腹股沟等部位皮肤粗糙,皮棘稍隆起,呈天鹅绒样外观(1B)。腹部皮损组织病理(图2):表皮角化过度,棘层肥厚,基底层可见色素沉着,真皮浅层扩张毛细血管周围少量慢性炎症细胞浸润。头颅MRI(图3):横断面T2WI示双侧脑室扩大,双侧额部蛛网膜下腔略增宽;矢状面T2WI示小脑扁桃体上下径增宽,下缘尖锐,超过枕骨大孔0.3 cm。符合Chiari畸形伴梗阻性脑积水。诊断:CAN。本研究通过福建医科大学附属第一医院医学伦理委员会批准,患儿监护人及正常人均签署知情同意书。
2.提取基因组DNA:分别抽取患儿及患儿父母、100例无亲缘关系的健康人静脉血各3Ml,放置于5Ml乙二胺四乙酸抗凝管中,-80℃储存,并用DNA提取试剂盒抽提基因组DNA。紫外分光光度计及琼脂糖凝胶电泳测定DNA浓度及纯度。
3.引物设计及PCR扩增:从数据库(https://www.ncbi.nlm.nih.gov)获得FGFR3基因组序列,应用Primer 5.0软件设计针对FGFR3基因的引物,正向引物:5′⁃TGGCGTTACTGAC TGCGAGA⁃3′,反向引物:5′⁃GTTTCGTGCCCCAAAGTACC⁃3′;片段长度521 bp。引物由生工生物工程(上海)股份有限公司合成。以患儿及其父母基因组DNA为模板,用Ex Taq聚合酶等PCR试剂(大连Takara公司)扩增FGFR3基因的外显子及其侧翼序列。PCR体系总体积20μl,含模板DNA 50 ng、正反向引物(20μmol/L)各0.5μl、Ex Taq Mix 10μl,加ddH2O补至20μl。PCR扩增条件:95℃预变性3min,95℃变性30 s,59℃退火30 s,72℃延伸45 s,40个循环后72℃延伸7min。
4.DNA测序:PCR产物经回收、纯化后用美国Applied Biosystems公司ABI3730XL型全自动测序仪测序,Chromas V2.23软件比对分析测序数据。用Chromas软件(版本2.0)分析基因序列,确定突变位点。
二、结果
患儿FGFR3基因第10外显子有1个杂合错义点突变(C.1172 C>A),导致该位点编码的氨基酸由丙氨酸变为谷氨酸(P.Ala391Glu);患儿父母及100例无血缘关系的健康对照中未发现该突变。见图4。
三、讨论
CAN由FGFR3基因突变引起[2],FGFR3的不同致病性突变可引起不同临床表现。有报道FGFR3 p.Lys650Thr突变可导致黑棘皮病改变,而无明显颜面和骨骼异常;p.Lys650Gln突变可导致季肋发育不全伴黑棘皮病;p.Ser348Cys突变导致中等程度软骨发育不全和黑棘皮病[3]。我们检测到该例患儿存在FGFR3基因1172位核苷酸C>A替换突变,使该位点所编码的氨基酸由丙氨酸变为谷氨酸(Ala391Glu),该突变可能是导致CAN的原因。FGFR3基因编码的成纤维细胞生长因子3是一种跨膜受体酪氨酸激酶,参与调节细胞生长、凋亡、分化、增殖、迁移和血管再生以及伤口愈合过程。FGFR3可能通过参与良性表皮细胞的生长及角质形成细胞增殖导致黑棘皮病[3]。有研究显示,FGFR3的A391E基因突变导致细胞表面活性突变受体表达减少,弱化信号传递,从而引起临床表型的发生[4]。
临床上CAN除具有Crouzon综合征的一般症状外,还可有一些特征性的变化,如后鼻孔闭锁、脑积水、胼胝体和小脑蚓体发育不全[5]等。骨骼表现包括牙骨质瘤和牙骨质发育不良。此外,还可能有软骨发育不全的特征,如小的坐骨切口、短指等[6]。CAN患者表现为泛发的棕褐色,天鹅绒样色素过度沉着,在身体皱褶部位表现更为明显,如口周、眼周、腋下、颈部。黑棘皮病的症状往往表现较早,通常在青春期前就表现出来,其严重程度有一定的差异[1]。有报道出生后即有黑棘皮病表现[7]。本文患儿出生时伴有颅面畸形,渐出现脑积水症状,影像学检查符合Chiari畸形伴梗阻性脑积水。患儿未做脑脊液引流,随访至今,智力发育正常,可能和颅内压升高不明显、未对神经系统造成明显损害有关。患儿还出现牙列不整、参差不齐、牙骨质发育不良和泪管堵塞,国外未见类似报道。患儿出现黑棘皮病的表现早,渐蔓延扩散,以皱褶及眶周明显,较普通的黑棘皮病受累广泛。

图1 患儿临床表现 头颅呈方形,前额突出,上颌发育不全,下颌前凸;眼球突出,双眼间距较宽;鼻梁塌陷,上下牙咬合不良(1A)。全身皮肤黑褐色,以颈部及腋下、腹股沟等部位明显,皮肤粗糙,皮棘稍隆起,呈天鹅绒样外观(1B)

图2 腹部皮损组织病理 表皮角化过度,棘层肥厚,基底层可见色素沉着,真皮浅层扩张毛细血管周围少量慢性炎症细胞浸润(HE×100)
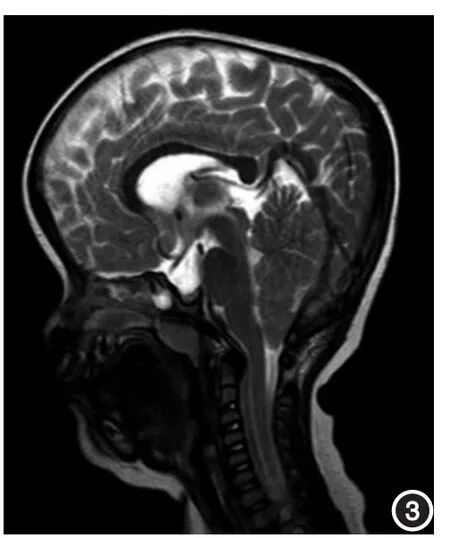
图3 头颅MRI 矢状面T2WI示小脑扁桃体上下径增宽,下缘尖锐,超过枕骨大孔0.3 cm

图4 患儿及其父母测序图 患儿携带c.1172C>A杂合突变位点(箭头所示);患儿父亲、母亲及健康人未显示突变
临床上,大多数CAN患者在出现黑棘皮病表现前未做基因检测,仅能做出Crouzon综合征的诊断,明确诊断需要结合FGFR3突变情况。CAN被认为是常染色体显性遗传,本文家系中仅患儿出现FGFR3基因突变,其父母及健康对照均未发现此突变,考虑为新发突变,与既往报道一致[8]。
猜你喜欢
娃娃乐园·综合智能(2022年9期)2022-08-16
上海金属(2021年6期)2021-12-02
昆明医科大学学报(2021年3期)2021-07-22
科学大众(2021年9期)2021-07-16
烟草科技(2021年6期)2021-06-24
中国生殖健康(2020年2期)2021-01-18
电脑知识与技术(2018年19期)2018-11-01
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
中国调味品(2017年2期)2017-03-20
