
Fe3O4羧甲基纤维素钠包覆量对酸性大红GR降解影响的研究
2018-08-04 01:18张鑫宇周益名沈娟莉薛国新
造纸化学品 2018年3期
张鑫宇,周益名,沈娟莉,盛 超,薛国新
(浙江理工大学材料与纺织学院·丝绸学院 制浆造纸研究所,浙江,杭州 310018)
当今,偶氮染料在油墨、造纸、印染、纺织等行业中大量使用,导致工业废水排放量巨大,是公认的主要环境污染源之一。高级氧化技术中的芬顿技术是目前工业废水处理中常用的方法。近些年,Gao等人报道Fe3O4MNPs具有类似于在天然过氧化物酶中发现的内在的酶的模拟活性,其在废水处理中被广泛用于氧化有机基质或作为检测工具[1]。有机配体可以与异相催化剂表面的金属离子发生络合反应,进而改变催化剂表面的物理化学性能。Niu等人证实腐殖酸涂覆的Fe3O4MNPs(Fe3O4/HA)高效分解H2O2生成大量的·OH[2]。Fe3O4/HA催化性能的提高与粒径、表面积和水分散性有关。Matta等人在不同有机配体络合的催化剂活化H2O2氧化三硝基甲苯的研究中指出:羧甲基纤维素(CMC)和乙二胺四乙酸(EDTA)有机配体的加入都能增强Fe3O4对H2O2的活化,其中CMC具有更好的增强效果[3]。在本课题组此前的研究也表明羧甲基纤维素钠包覆后的Fe3O4对酸性大红GR的氧化降解具有良好的催化效果[4]。
本研究主要探究了Fe3O4羧甲基纤维素钠包覆量对其催化效率的影响,制备3种不同羧甲基纤维素钠包覆量的Fe3O4的催化剂,通过实验对比其催化效率,选定比较优质的催化剂,并对其在芬顿体系中降解酸性大红GR的机理进行了分析。
1 实验
1.1 仪器与试剂
仪器:JY-D2-ⅡN超声仪细胞粉碎机、RigakuD/MAX-200X射线衍射仪、Nicolet 5700傅里叶红外光谱仪、722 s可见分光光度计、Acquity UPLLC高效液相色谱、JSM 7610F扫描电子显微镜。
实验使用主要试剂:羧甲基纤维素钠、无水乙醇、过氧化氢、氢氧化钠、盐酸、四水氯化亚铁、六水氯化铁、酸性大红GR,购自杭州米克化工仪器公司。
1.2 Fe3O4/CMC的制备
实验中制备催化剂的具体方法为:通过化学共沉淀法制备复合材料Fe3O4/CMC[5]。简言之,将2.0 g四水氯化亚铁(FeCl2·4H2O)和5.2 g的六水氯化铁(FeCl3·6H2O)溶解于100 mL去离子水中,将溶液倒入三口烧瓶中放入恒温水浴锅中进行90℃恒温水浴,三口烧瓶中的溶液在N2保护下持续搅拌。然后加入羧甲基纤维素钠(CMC-Na)(0.1、0.3、0.5 g)分别溶于50 mL去离子水中配制成的溶液和10 mL氨水(25%)快速依次加入到反应溶液中。将混合物陈化反应30 min,然后用凉水冷却至室温。使用强力磁铁从溶液中分离得到的磁性纳米颗粒,并用去离子水反复清洗3次后干燥,最终得到Fe3O4/CMC复合材料(根据Fe3O4包覆CMC-Na量的不同,分别命名为1号、2号和3号复合催化剂Fe3O4/CMC,以下简称1号、2号和
3号催化剂)。
1.3 反应溶液的分析及检测方法
根据郎伯-比尔定律,一定条件下溶液吸光度与溶液浓度成正比。本实验中酸性大红GR使用可见分光光度计在510 nm波长进行测试,使用式(1)即可计算出酸性大红GR的降解率。

式中:A0为染液初始吸光度;A为染液处理后吸光度。
2 结果与讨论
2.1 复合材料的表征
图1为3号催化剂的扫描电镜照片。

图1 3号催化剂的扫描电镜照片
从图1可以看出,所制备的催化剂为50 nm左右的球形颗粒。即可认为本研究中制备的复合材料为纳米级别的粒子。
图2显示了实验所制备的1号、2号和3号催化剂(分别用“1”、“2”和“3”表示,下同)的 X 射线衍射谱图。
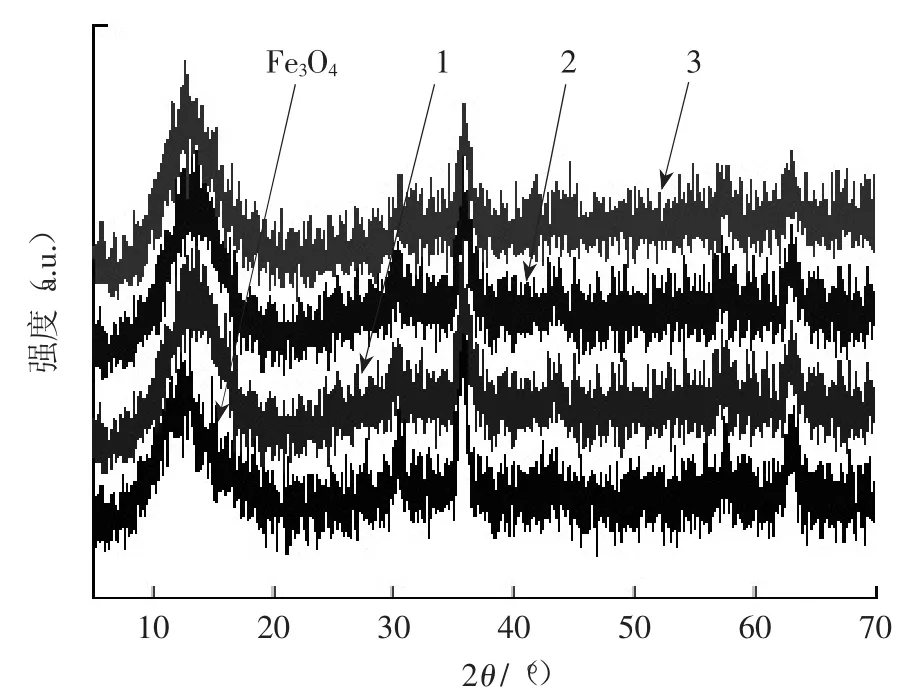
图2 1号、2号、3号催化剂的X射线衍射谱图
图谱中在 5°~70°之间有 30.2°、35.6°、42.3°、53.6°、57.1°和 62.6°6 个特征衍射峰,分别对应的晶面是(220)、(311)、(400)、(422)、(511)和(440),与标准谱图对比可知,这是Fe3O4尖晶石的6个晶面,说明自制的复合材料仍具有Fe3O4尖晶石型的晶型结构。
图3为1号、2号和3号催化剂的红外光谱图。
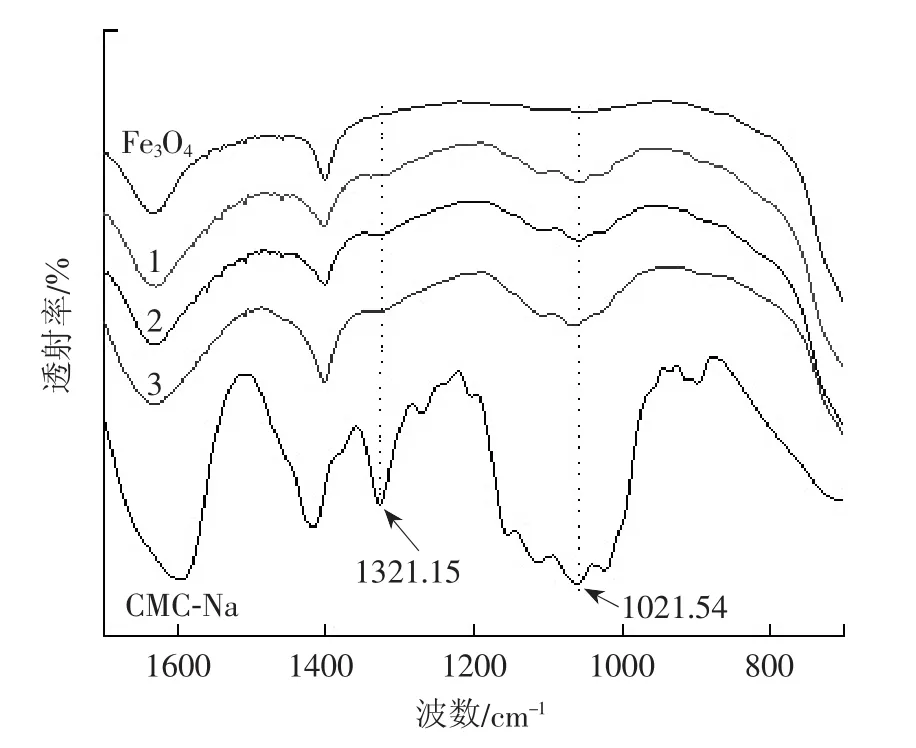
图3 1号、2号和3号催化剂的红外光谱图
如图3所示,通过红外光谱分析1号、2号、3号催化剂和CMC-Na。CMC-Na在1 321.15 cm-1和1 021.54 cm-1有2处特征峰,在1 321.15 cm-1处的特征峰是由于—OH的弯曲振动,1 021.54 cm-1处特征峰是由于其结构上CH—O—CH2基团的拉伸[6-7]。制备的1号、2号和3号催化剂实验样品在相同位置处也存在特征峰,但Fe3O4在这2处位置没有特征峰,说明1号、2号和3号催化剂样品中既含有Fe3O4又包含一定量的CMC-Na,即CMC-Na已成功修饰到Fe3O4上。
2.2 非均相类芬顿体系降解酸性大红GR的研究
通过改变酸性大红GR初始浓度、催化剂投量及反应溶液pH制备不同类型的反应液,然后100 mL反应溶液加入一定量H2O2,最后将样品放入40℃的恒温空气浴振荡器中,每隔一段时间进行取样测试,一共测试12次。
2.2.1 1号、2号和3号催化剂对酸性大红GR降解率的对比
本实验采用酸性大红GR作为目标底物在同一实验条件(催化剂投加量为0.5 g/L,H2O2的量为20 μL,酸性大红GR质量浓度为50 mg/L,pH=3.5)下,对本研究制备的1号、2号和3号催化剂进行催化实验对比,所得结果见图4。

图4 1号、2号和3号催化剂对酸性大红GR的降解率对比
由图4可知,在相同条件下3号催化剂对酸性大红GR的降解作用最佳,反应3 h后,使用3号催化剂的反应体系酸性大红GR的降解率达到了90%以上。3号催化剂相对于1、2号催化剂,其包覆的羧甲基纤维素钠量最多,在水溶液中具有更好的分散性能,能够减少纳米粒子的团聚,暴露出的活性位点更多,性质更好,因而在类芬顿体系中具有更好的催化作用。
2.2.2 pH对酸性大红GR降解率的影响
在类芬顿体系中,pH是影响反应效率的重要影响因素之一。在3号催化剂投加量为0.05 g/L,H2O2的量为20 μL,酸性大红GR质量浓度为50 mg/L的实验条件下,3号催化剂构建的非均相类芬顿体系中pH与酸性大红GR降解率的关系如图5所示。

图5 初始pH对酸性大红GR降解率的影响
从图5看出:在6 h的反应时间内溶液中酸性大红GR降解效率大致随着反应初始pH的增大而减小;当pH大于等于5.5时酸性大红GR氧化降解的速度比较慢,反应6 h后仅仅只有不到10%的降解率;当调节酸性大红GR溶液pH=4.5时,开始1.5 h酸性大红GR的降解速度较为缓慢,1.5 h后降解速度大幅提高,反应6 h后达到83%的降解率;当调节酸性大红GR溶液pH=3.5时,酸性大红GR的降解效率大幅提高,反应1.5 h后即可达到90%的降解率。当调节酸性大红GR溶液pH=3.0时,酸性大红GR的降解效率也出现了一定幅度的降低。同样是由于反应体系中自由基与H+进行反应,降低了反应溶液中自由基的浓度,进而影响了酸性大红GR的降解效率。
2.2.3 H2O2浓度对酸性大红GR降解效率的影响
在3号催化剂投加量为0.05 g/L,pH=3.5,酸性大红GR质量浓度为50 mg/L的实验条件下,3号催化剂构建的非均相类芬顿体系中H2O2浓度对酸性大红GR降解率的影响如图6所示。

图6 H2O2浓度对酸性大红GR降解率的影响
从图6可以看出,当反应时间进行1 h时,投放10 μL H2O2条件下的实验组酸性大红GR也达到了70%氧化降解率。当H2O2的投放量增加到40 μL时,体系中的酸性大红GR在2 h内可以达到92%的降解率。大致上看,反应体系中随着H2O2浓度的升高酸性大红GR的降解效率越高、降解效果越好。
从图6也可以看到特例,实验过程中投加的H2O2为50 μL时体系的催化降解效率稍弱于投放量为40 μL的情况,文献中一些类似报道,主要原因为H2O2浓度过高时体系中H2O2与自由基的会发生类似于式(2)的反应[8-9]:

适量增加H2O2的投加量可以小幅提高体系中酸性大红GR的降解率,但不能过量添加,过量添加一方面不仅不会提高酸性大红GR的降解效率,反而会导致降低酸性大红GR的降解效率降低,另一方面
还会造成H2O2的浪费,增加了使用成本。
2.2.4 3号催化剂投加量对酸性大红GR降解效率的影响
在 H2O2的量为 20 μL,pH=3.5,酸性大红 GR 质量浓度为50 mg/L的实验条件下,3号催化剂投加量对酸性大红GR降解率的影响如图7所示。

图7 3号催化剂投加量对酸性大红GR降解率的影响
由图7可知:在3号催化剂最少投放量0.010 g/L的条件下,体系反应3 h时后,溶液中的酸性大红GR已经达到了90%的降解率;增加投放量到0.25 g/L时,反应速度小幅提高,2.5 h可达到90%左右的去除率;当反应体系中自制催化剂的投加量分别增加到0.050、0.075和0.100 g/L时,反应速率也大为提高,1.5 h即可达到90%左右的去除率反应过程十分相近,3条过程反应曲线几乎重合。这与使用的催化剂有着很大的关系,3号催化剂在制备过程中只用了较多的CMC-Na,制备出来的催化剂分散性最好。催化剂的分散性越好,暴露出来的活性位点越多,活性位点的增加就使类芬顿体系中的酸性大红GR降解效率越高。
2.2.5 底物酸性大红GR初始浓度对降解效率的影响
在 H2O2的量为 20 μL,pH=3.5,3 号催化剂投加量为0.05 g/L的实验条件下,底物酸性大红GR初始浓度对降解率的影响如图8所示。

图8 酸性大红GR的初始浓度对降解率的影响
从反应得到的五组曲线的对比中可以发现:有机物底物酸性大红GR的降解速率随着初始浓度的增大而降低,虽然当酸性大红GR初始质量浓度降低至10 mg/L时,有机物的转化又产生了效率低下的现象,但是反应开始的1.5 h内仍可保证接近于90%的去除率;当酸性大红GR初始质量浓度大于、等于25 mg/L小于等于75 mg/L时,反应过程中酸性大红GR的去除率比较接近,特别是1 h后的降解趋势十分相似,在反应开始的1 h内都达到了高于60%的转化率。而当酸性大红GR的初始质量浓度增加至100 mg/L后,反应速率最低,即便如此,5组反应于2.5 h后的降解情况几乎一致。
2.2.6 催化剂的可回收性研究
对3号催化剂进行了多次回收利用实验,每次实验反应结束后使用磁铁将溶液中的自制催化剂进行回收,回收后的催化剂使用去离子水简单清洗后再次在上述条件下进行使用,测定酸性大红GR残余浓度后得到图9(实验条件:催化剂投加量0.5 g/L,H2O2量为20 μL,酸性大红GR质量浓度为50 mg/L,pH=3.5,温度为40℃)。

图9 催化剂连续重复利用4次的催化效果
从图9可以看出,第2次回收的催化剂在反应6 h后将酸性大红GR催化氧化为原浓度14.8%。第3次回收的浓度只有第一次使用时的29.5%,第3次使用对比第1次使用,其催化效率明显降低,反应后溶液的酸性大红GR浓度为原溶液的35.1%。同样现象出现在之后的重复利用中,催化剂的催化作用明显衰弱。推测以上现象产生的原因主要是酸性大红GR及中间产物等副产物对自制复合催化剂上活性点位具有一定的毒性作用,其次是自制复合催化剂在反应体系中结构遭到破坏,分散性能变差,导致在水溶液中暴露的活性位点减少[10-11]。
2.3 非均相类芬顿体系中的反应历程研究
2.3.1 电子自旋共振波谱(EPR)
在上一节中进行了酸性大红GR的氧化降解实验,为了说明非均相类芬顿体系中酸性大红GR的降解是由于自制复合催化剂Fe3O4/CMC催化H2O2产生·OH,故此进行了·OH捕获实验,即EPR测试,测试结果如图10所示。

图10 EPR谱·OH自由基
图10给出了5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)捕获自由基的EPR图谱。由该谱图可知,在H2O2-Fe3O4/CMC体系中,出现了强度比为1∶2∶2∶1的DMPO-OH附加物四频特征峰[12],由此说明了体系中·OH的存在,进一步说明了非均相类芬顿体系中酸性大红GR的降解是由于Fe3O4/CMC催化H2O2产生·OH自由基,进而被·OH氧化降解的。
2.3.2 Fe3O4/CMC催化H2O2作用机理的分析
在酸性条件下,催化剂表面的二价铁活性点位上可以与H2O2作用产生·OH如式(3),催化剂表面的三价铁亦可与H2O2作用被还原为Fe2+如式(4):

图11为反应溶液的颜色变化,开始时溶液为红色,反应2 h后为黄色,反应结束后为无色。

图11 酸性大红GR溶液(a)、降解2 h后溶液(b)反应后溶液(c)的颜色变化
3 结论
本实验成功制备出1号、2号和3号Fe3O4/CMC复合材料,以此作为非均相类芬顿体系中的催化剂进行酸性大红GR的氧化降解实验。对比发现,在相同的反应条件下,3号Fe3O4/CMC催化剂对酸性大红GR催化氧化降解的效率最高,即Fe3O4包覆羧甲基纤维素钠的增加,催化效果也会提高。同时催化剂在循环使用4次后,仍然具有良好的催化能力。通过EPR测试,在自由基捕获剂的作用下检测出·OH,自制催化剂可以催化过氧化氢产生强氧化性的·OH,进而将溶液中的酸性大红GR氧化降解。
猜你喜欢
城市道桥与防洪(2022年5期)2022-06-25
包装学报(2022年2期)2022-05-13
造纸化学品(2019年6期)2020-01-17
福建基础教育研究(2019年8期)2019-05-28
天然产物研究与开发(2019年1期)2019-03-01
中国资源综合利用(2017年2期)2018-01-22
中国资源综合利用(2017年2期)2018-01-22
中国造纸学报(2017年4期)2018-01-03
中学生数理化·高二版(2016年3期)2016-12-26
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18
