
儿童皮下脂膜炎样 T 细胞淋巴瘤 1 例病例报告
2018-08-03 05:05李艳华廖雪莲蒋莎义
中国循证儿科杂志 2018年3期
李艳华 廖雪莲 蒋莎义 蒋 慧
1 病例资料
女,9 岁,2016 年4月 2日因“发热 1月余伴臀部软组织肿胀”于上海市儿童医院(我院)住院治疗。患儿的重要临床信息见图1。
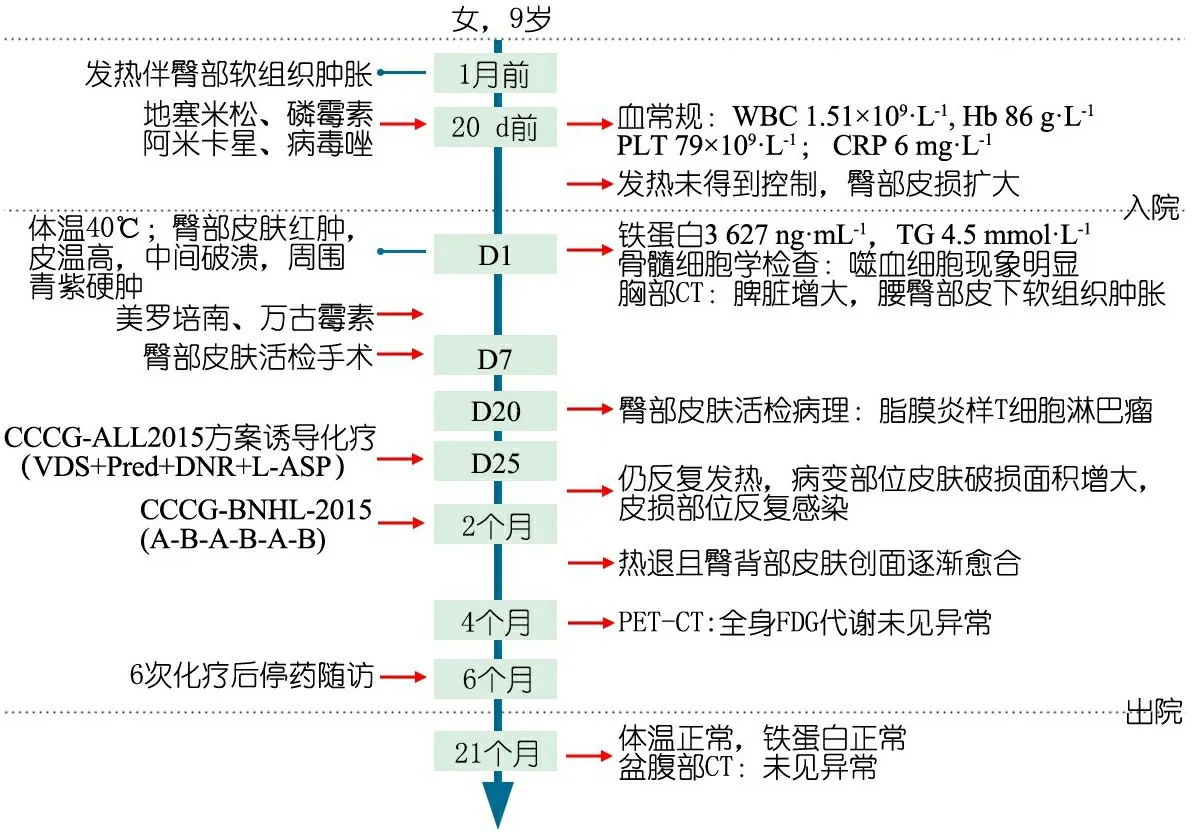
图1 本文病例重要临床信息时间轴图
患儿 1 个月前出现发热,呈持续高热,热峰>39℃,并出现臀部软组织肿胀,皮肤呈青紫状,肿胀范围渐扩大,中间皮肤破溃,无渗液,无触痛,无髋关节活动障碍。外院曾予阿米卡星、磷霉素、病毒唑和地塞米松等,仍发热,臀部皮损扩大。患儿既往体健,生长发育正常,否认家族遗传病史。
体格检查:体温40℃,心率 122·min-1,呼吸 21·min-1,血压 94/68 mmHg。体重 38 kg(P90),身高 144 cm(P90)。神志清,反应可。双肺听诊无异常。臀部皮肤红肿,皮温高,中间皮肤破溃,周围青紫硬肿(图 2A)。右侧腹股沟处触及 1 枚肿大淋巴结,质地软。心肺未见异常,肝脾肋下未及。四肢肌力、肌张力可,关节无畸形、压痛,病理反射未引出。
辅助检查:血常规 WBC 1.51×109·L-1, Hb 86 g·L-1, PLT 79×109·L-1,CRP 6 mg·L-1。血生化检查显示,胆红素正常,ALT 正常,AST 130 U·L-1,LDH 1 714 U·L-1,白蛋白32.8 g·L-1,TG 4.5 mmol·L-1,铁蛋白 4 943 ng·mL-1。骨髓细胞学检查显示,噬血细胞现象明显。胸部CT两肺未见明显异常。颅脑MRI 和胸部 CT 未见异常。腹部 CT显示,脾脏增大,腰臀部皮下软组织弥漫性肿胀。我院病理报告(2016 年 4 月 7 日):右侧臀部皮肤活检(图3),镜下见皮肤附件周围及皮下脂肪组织中大量淋巴细胞和组织细胞浸润,可见核碎屑,呈脂膜炎样改变,需除外脂膜炎样 T 细胞淋巴瘤;免疫组化Vim+,CD3+,CD20少量表达,CD1a-,S100少量表达, CD21-,ALK-,CD30部分表达,CD15-, Ki-67+80%,CD8+,TIA-1 部分表达。腹股沟淋巴结活检,镜下见淋巴窦内大量组织细胞样细胞增生,淋巴结副皮质区增生,以中小淋巴细胞和组织细胞增生为主,考虑淋巴组织反应性增生,需除外淋巴瘤。TCR基因重排结果α/β+。2016 年 4 月 23 日复旦大学附属肿瘤医院病理科臀部皮肤活检提示,结合形态、免疫组化和原位杂交结果,符合脂膜炎样 T 细胞淋巴瘤;免 疫 组 化 ( HI16-6540 ) : 肿 瘤 细 胞CD3+,CD7+,CD56-,granzymeB 部 分+,perforin+,CD20-,CD30-,Ki-67+70%,组织细胞 kpl+。原位杂交:EB病毒阳性。腹股沟淋巴结淋巴组织增生,窦组织细胞增生;免疫组化(HI16-6540):生发中心 CD20+, 副皮质区小淋巴细胞 CD3+,组织细胞 kpl+,CD30-。
结合患儿的临床表现及病理结果,诊断为皮下脂膜炎样 T 细胞淋巴瘤(SPTCL)合并噬血细胞综合征。2016 年 4 月 27 日至5 月 29 日予 CCCG-ALL2015 方案诱导化疗[长春地辛(VDS)+醋酸泼尼松片(Pred)+柔红霉素(DNR)+门冬酰胺酶(L-ASP),33 d)],同时予对症治疗。患儿仍反复发热,病变部位皮肤破溃面积增大,皮损部位反复感染。2016 年 6 月 6 日调整化疗方案为 CCCG-BNHL-2015(A-B-A-B-A-B ,A:长春地辛、泼尼松、环磷酰胺、表柔比星、阿糖胞苷;B: 长春地辛、泼尼松、异环磷酰胺、依托泊苷、甲氨喋呤),每个疗程间隔3周左右。完成2 个疗程后体温正常,臀背部皮肤创面逐渐愈合。2016 年 7 月 28 日全身 PET-CT 检查报告“全身未见肿大淋巴结,FDG 代谢未见异常,右侧腹股沟软组织肿胀,考虑穿刺后改变,脑 FDG 代谢未见异常”。化疗6 个疗程后,臀背部皮肤创面完全愈合(图 2B)。2017 年 3 月 13 日全身 PET-CT 检查报告“鼻咽部、咽周淋巴环、颈部淋巴结 FDG 代谢升高,建议密切随访除外淋巴瘤侵犯, 余未见明显异常”。2017年3 月 20 日五官科行鼻咽部活检病理示炎性改变。停药随访 15 个月, 处于完全缓解状态。
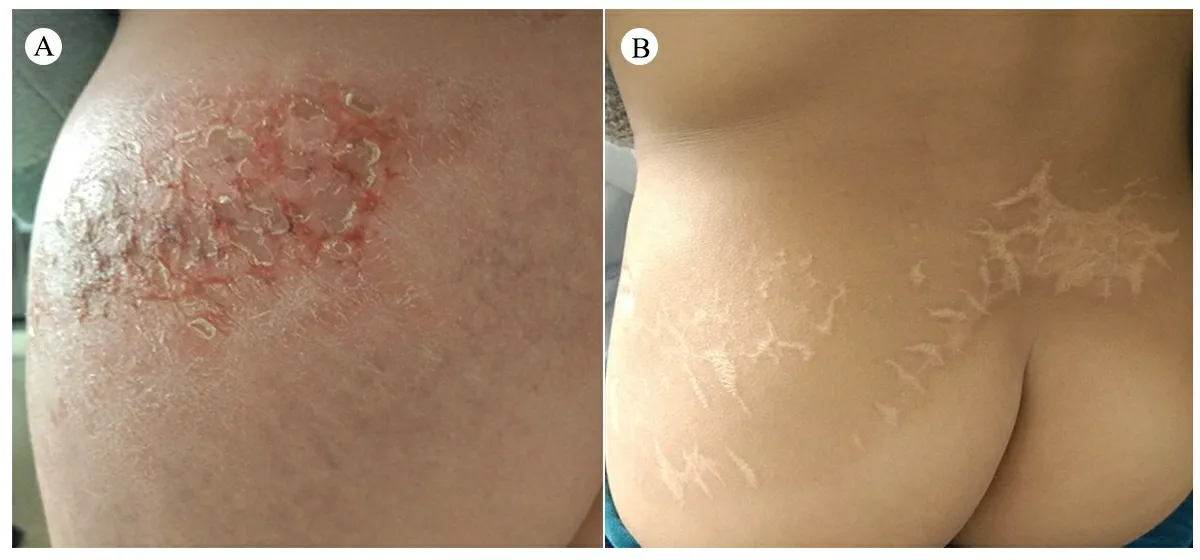
图2 患儿治疗前后皮损表现
注 A :治疗前,臀部及腰部皮肤红肿,皮温高, 中间质地软、皮肤破溃,周围青紫硬肿;B:治疗后,臀部及腰部皮肤无红肿、创面愈合
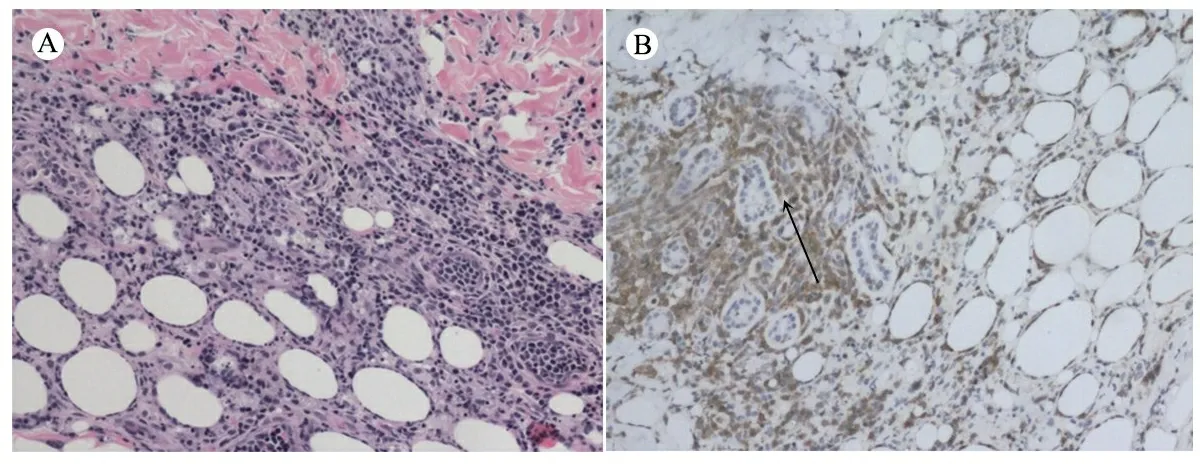
图3 患儿臀部皮肤活检病理表现
注 A:HE,×100,异型的大小不一的肿瘤细胞浸润脂肪组织和附属器周边,肿瘤细胞在单个脂肪细胞周边围绕,可见核碎屑;B:IHC,×100,肿瘤细胞表达 CD3,细胞浆着色
2 讨论
SPTCL是指主要累及皮下脂肪组织且与脂膜炎相似的一种原发于皮肤的外周 T 细胞淋巴瘤,是近年来确定的一种皮肤原发淋巴瘤的新亚型[1],占所有外周 T 细胞淋巴瘤的 0.5%~0.9%[2],主要见于成人,儿童少见。SPTCL 以皮损和皮下结节为主要表现,好发于肢体,其次为躯干,也可累及面、颈、踝、腋窝、腹股沟和臀部,呈多部位发生的黄褐色、皮下无痛性结节或斑块,亦有似蜂窝组织炎样改变[3];早期可无明显淋巴结受累;可累及多系统,合并噬血细胞综合征[4]。本文患儿臀部皮损经病理检查明确诊断为SPTCL,并继发噬血细胞综合征。
以“Subcutaneous panniculitis like T cell lymphoma”为关键词检索PubMed和万方数据库,检索时间为建库至 2018 年3月1日,共检索到37篇文献报告45例SPTCL患者,国内11例,国外34例,与本病例合并后共46例,平均发病年龄为28岁。其中儿童 14 例(30%),发病年龄8个月至17岁(中位年龄10.5 岁)。46 例SPTCL均出现皮损、结节,1 例表现为眼眶蜂窝组织炎(2%),14 例有脏器浸润(30%),18 例继发噬血细胞综合征(39%)。本文患儿发病年龄 9 岁,主要表现为臀背部皮损,继发噬血细胞综合征,无脏器累及。
SPTCL 临床表现差异较大,诊断主要依靠活组织病理检查,表现为肿瘤细胞浸润局限于皮下组织,极少侵犯真皮深部,肿瘤细胞多形、核大、形态不规则、染色质致密,浸润于脂肪细胞之间,呈花边样方式,常见脂肪细胞坏死、核碎裂,病变严重时脂肪细胞可广泛坏死。脂肪坏死常导致组织细胞反应,包括多核巨细胞或肉芽肿,巨噬细胞单个散布于肿瘤细胞之间,与肿瘤细胞相混合,常吞噬红细胞、中性粒细胞、血小板或核碎片。有时部分区域可见出血。虽可见血管浸润,但无血管中心侵袭或破坏[5]。免疫组化和流式细胞检测均显示表达异常的 T 细胞亚群 CD2、CD3、CD5、CD7、CD8 阳性和 CD20、CD34 阴性,不表达 B 细胞相关抗原,大部分病例的皮损中可检测到TCR基因重排,检测不到免疫球蛋白重或轻链基因重排,说明肿瘤细胞来源于 T 细胞[6]。根据TCR基因重排,SPTCL 被分为α/β亚型和γ/δ亚型,两种亚型的临床表现、治疗和预后均不同[6]。α/β亚型患者病变多局限于皮下组织,通常 CD8 阳性、CD56阴性,临床病程相对缓和,预后较好。文献复习的46 例患者均经病理检查诊断为 SPTCL,其中 24 例检测到α/βTCR基因重排(52%),16 例基因重排情况不详(34.7%)。γ/δ亚型肿瘤细胞多表达 CD56,合并噬血细胞综合征,患者病情进展迅速,多在短时间内死亡,预后极差,2018 年欧洲癌症组织(WHO-EORTC)将其定义为皮肤γ/δT 细胞淋巴瘤(PCGD-TCL)。本文患儿中,肿瘤细胞表达 CD3和CD8,α/βTCR基因重排阳性。
SPTCL缺乏特异性临床表现、病理特征和统一的诊断标准,早期诊断困难,易误诊、误治,有文献报道,SPTCL发病至确诊平均时间 28.6 个月[7],文献复习的46 例患者的平均诊断时间为 10 个月。WHO-EORTC统计,SPTCL的5 年生存率为 82%,15%~20%患者合并噬血细胞综合征,5 年生存率降至50%[8]。46 例中死亡10 例(21.7%),其中 5 例继发噬血细胞综合征,1 例存在骨髓浸润,3 例肿瘤细胞表达 CD56+。18 例继发噬血细胞综合征患者中,6例缓解(44.4%),4例失访(22.2%),5例死亡(16.7%),3例(16.7%)化疗后复发,行造血干细胞移植后缓解。目前认为,继发噬血细胞综合征、肿瘤细胞 CD56阳性和多脏器受累为SPTCL的预后不良因素[9]。
国内外对 SPTCL 治疗方案迄今尚未达成一致,既往文献报道中多采用联合化疗,以 CHOP 方案为主[10-12]。46 例中2 例(4.3%)临床表现为皮损、未继发噬血细胞综合征、CD56阴性,未经治疗存活;8例(17.4%)以口服环孢素治疗为主;16 例(34.8%)予 CHOP方案化疗,9 例缓解,7 例疾病进展或复发,7例中的4例患者行造血干细胞移植后缓解;5例(11%)放弃治疗,5例化疗方案不详(11%),1例(2%)SMILE方案化疗,2例(4.3%)罗米地辛化疗,余7例(15%)为环磷酰胺、阿糖胞苷、依托泊苷化疗。目前认为,造血干细胞移植对化疗无效的α/β SPTCL 患者有重要治疗价值[13-15]。本文患儿病初即合并噬血细胞综合征,存在预后不良高危因素,予 CCCG-BNHL-2015 方案化疗后,疾病完全缓解。
综上所述,SPTCL的临床表现差异大,容易误诊,诊断须以病理学检查为基础。SPTCL 患者合并噬血细胞综合征,提示预后不良,本文病例予 CCCG-BNHL-2015 方案化疗后疾病完全缓解,为 SPTCL 治疗提供了新的参考方案,但本病例诊断时无脏器受累,且肿瘤细胞 CD56阴性,这些因素可能与良好预后相关。
猜你喜欢
传染病信息(2022年3期)2022-07-15
中国药学药品知识仓库(2022年9期)2022-05-23
锦州医科大学报(2022年2期)2022-05-07
现代临床医学(2021年2期)2021-03-29
河南科学(2020年3期)2020-06-02
自我保健(2020年1期)2020-03-13
散文诗世界(2019年6期)2019-09-10
中国临床医学影像杂志(2019年6期)2019-08-27
中国临床医学影像杂志(2019年5期)2019-08-27
家庭科学·新健康(2017年12期)2018-01-09
