
Gene expression during different periods of the handlingstress response in Pampus argenteus*
2018-08-02 02:51:08SUNPeng孙鹏TANGBaojun唐保军YINFei尹飞
SUN Peng (孙鹏), TANG Baojun (唐保军), YIN Fei (尹飞)
Key Laboratory of East China Sea Fishery Resources Exploitation, Ministry of Agriculture; East China Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Shanghai 200090, China
Abstract Common aquaculture practices subject fish to a variety of acute and chronic stressors.Such stressors are inherent in aquaculture production but can adversely affect survival, growth, immune response, reproductive capacity, and behavior. Understanding the biological mechanisms underlying stress responses helps with methods to alleviate the negative effects through better aquaculture practices, resulting in improved animal welfare and production efficiency. In the present study, transcriptome sequencing of liver and kidney was performed in silver pomfret ( Pampus argenteus) subjected to handling stress versus controls. A total of 162.19 million clean reads were assembled to 30 339 unigenes. The quality of the assembly was high, with an N50 length of 2 472 bases. For function classi fication and pathway assignment,the unigenes were categorized into three GO (gene ontology) categories, twenty-six clusters of eggNOG(evolutionary genealogy of genes: non-supervised orthologous groups) function categories, and thirty-eight KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways. Stress affected different functional groups of genes in the tissues studied. Differentially expressed genes were mainly involved in metabolic pathways(carbohydrate metabolism, lipid metabolism, amino-acid metabolism, uptake of cofactors and vitamins, and biosynthesis of other secondary metabolites), environmental information processing (signaling molecules and their interactions), organismal systems (endocrine system, digestive system), and disease (immune,neurodegenerative, endocrine and metabolic diseases). This is the first reported analysis of genome-wide transcriptome in P. argenteus, and the findings expand our understanding of the silver pomfret genome and gene expression in association with stress. The results will be useful to future analyses of functional genes and studies of healthy arti ficial breeding in P. argenteus and other related fish species.
Keyword: gene ontology; immune system; next-generation sequencing; transcriptome; unigenes
1 INTRODUCTION
Cultured fish are routinely exposed to a variety of natural and arti ficial stressors (Campbell et al., 1992;Bonga, 1997; Øverli et al., 2005; Jia et al., 2016).Although the stress response is necessary for allostasis, particular aspects of the response may come at the expense of other physiological functions(i.e., adverse effects on growth, immune response,and reproduction) (Davis, 2006; Lefèvre et al., 2008;Harmon, 2009; Sahin et al., 2014). Gaining information on biological mechanisms of the stress response is helpful for aquaculture management, by improving the welfare of fish and ultimate production efficiency (Sánchez et al., 2011). Transcriptome pro filing has been an efficient approach in functional genomics and gene pro filing as relates to cells, tissues,organismal systems, or physiological conditions(Morozova and Marra, 2008; Wang et al., 2009). In the past decade, next-generation sequencing (NGS)technology has provided efficient methods for quantifying transcriptomes (Ozsolak and Milos, 2011;Parma et al., 2016). Studies of the stress response at the transcriptome level are helpful to ascertain the underlying genetic basis of adaptations. These studies represent a powerful way to evaluate the differential gene expression involved in an organism’s adaptation to a challenged or changed environment (Smith et al.,2013; Qian et al., 2016; Li et al., 2017).
As an economically and ecologically important fish species in China, the silver pomfret Pampus argenteus (Euphrasen 1788) has been studied since the 1980s (Huang et al., 2010). Because the species is sensitive to a variety of stressors, high mortality and low growth rates are often found in culture enterprises.By far, ongoing studies of P. argenteus are mainly involved with nutrition, physiology and reproduction(Huang et al., 2010; Gao et al., 2016), whereas information on genome organization and gene expression in Pampus species is still very limited. In the present study, de novo transcriptome sequencing in P. argenteus was performed using the Hiseq2000 platform. The project’s objective was to achieve deepsequencing of P. argenteus transcriptome and so provide a well-assembled transcriptome resource for a fuller understanding of the silver pomfret genome,and particularly changes in gene expression associated with handling stress. The transcriptome data obtained will also assist future studies of functional genes in Pampus species.
2 MATERIAL AND METHOD
2.1 Fish and experimental protocols
Silver pomfret P. argenteus were bred and cultured at the Zhoushan Aquatic Products Research Institute(Zhoushan, Zhejiang Province, China). Healthy fish were selected for the present study. Juvenile silver pomfret with a body weight of 32.16±1.1 g (mean±SD)were reared in 3000-L PVE tanks, with water temperatures maintained at 24–27°C. The experimental protocols were based on the work of Sun et al. (2017). Healthy fish were randomly divided into three groups (groups A, B and C), with each group kept in triplicate tanks (A1–A3, B1–B3, C1–C3), at 30 fish/tank. Group A fish were left undisturbed;fish from groups B and C were subjected to repeated handling stress by holding each fish individually in a net out of the water for 30 s, according to the method of Castanheira et al. (2013) with some modi fication.Before the stress treatment, 6 fish were sampled from group A as a control; visual inspection was employed every 2 h for the first 24 h, and every 4 h after 48 h, to identify possibly injured or dead fish. Six fish were separately sampled from both groups B and C, at 24 h post-stress and at 120 h post-stress, respectively. In addition, all tissues to be used for later analysis were sampled at almost the same time to minimize the effects of factors such as diurnal rhythm, photoperiod,and culture density. Fish were ethically euthanized in sea water containing a 100 mg/L concentration of MS-222 (Sigma); liver and kidney tissue samples were immediately collected and then frozen in liquid nitrogen for further processing.
2.2 RNA extraction and mRNA-seq library preparation for illumina sequencing platforms
Total RNA was extracted from the liver and kidney tissues using a TRIzol kit (Invitrogen, USA); the RNA samples were digested by RNase-free DNase I(5 U/μL), according to the manufacturer’s instructions.The concentration and integrity of the RNA samples were checked with a QuantiFluor®-ST Fluorometer(Promega) and Agilent 2100 Bioanalyzer (Agilent Technologies, USA). High-quality RNA samples from equal amounts of the two tissues were pooled for cDNA synthesis and sequencing, following the methods of Sánchez et al. (2011). A paired-end library was constructed from the cDNA, and short fragments with a desirable length were then puri fied using a QIAquick PCR Extraction Kit (Qiagen), end repaired,and linked with sequencing adapters. The Illumina Hiseq 2000 platform was used to generate sequence read pools (Shanghai Personal Biotechnology Co.,Ltd.).
2.3 Processing the sequence data, and de novo assembly
Before de novo assembly, raw sequencing reads were trimmed to remove adaptor, low-quality and short-read sequences, using the tool Cutadapt 1.2.1(http://code.google.com/p/cutadapt/) (Martin, 2011)and FastQC software. The assembly was completed using the de Bruijin graph approach (Grabherr et al.,2011; Haas et al., 2013).
2.4 Transcriptome annotation and gene ontology
The assembled contigs were used for searches with the Basic Local Alignment Search Tool (BLAST).The cut-off e-value was set at 1e-10. Gene ontology(GO) analysis was conducted using the zebra fish genome BLAST results in Blast2GO (http://www.BLAST2go.com). The GO annotation results were used to describe the associated molecular function,cellular component, and biological process. KEGG pathway maps were assigned using the online KEGG Automatic Annotation Server (http://www.genome.jp/kegg/tool/map_pathway2.html). Finally, analysis of the eggNOG (evolutionary genealogy of genes:non-supervised orthologous groups) distribution was conducted by online servers (http://eggnog.embl.de/version_3.0).
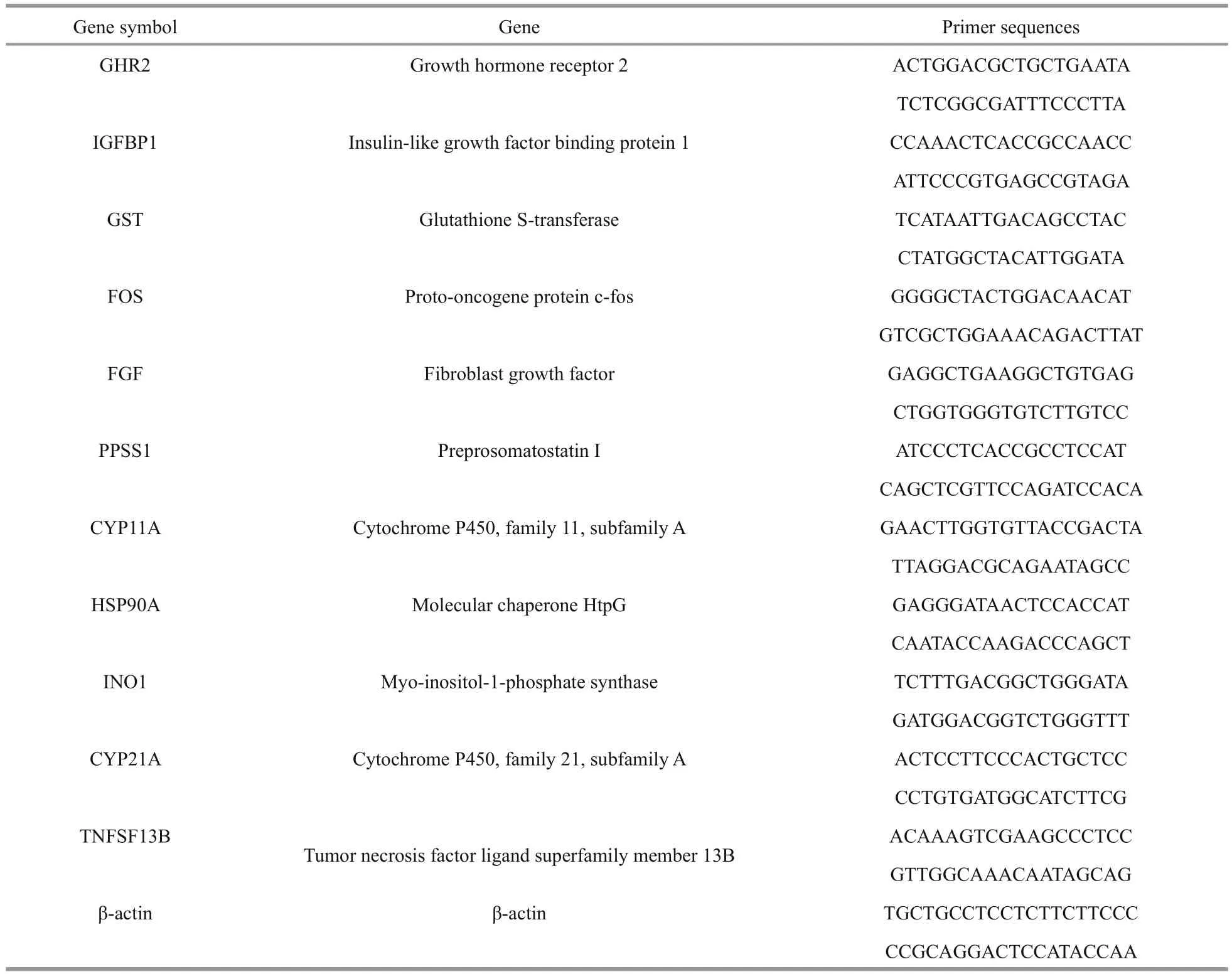
Table 1 Quantitative PCR gene and primer information
2.5 Identi fication of differentially expressed genes
Differentially expressed unigenes were analyzed by DESeq software (Anders and Huber, 2010). RPKM(reads per kilobase of exon model per million mapped reads) were used to normalize the estimates of transcript abundance (Mortazavi et al., 2008);identi fication of differential gene expression among different groups used 2-fold differentials.
2.6 Real-time PCR
Gene-expression analysis was validated and quanti fied by real-time PCR (RT-PCR). Primers for 11 genes involved with growth, metabolism, and immunity were designed by Premier 5 (Table 1). RNA extraction and cDNA synthesis were performed as described above. Reactions were optimized for the following genes: ghr 2, igfbp 1, gst, fos, fgf, ppss 1,cyp 11A, hsp 90A, ino 1, cyp 21A, and tnfsf 13B. RTPCR pro filing consisted of initial denaturing at 95°C for 5 min, followed by 40 cycles of denaturing at 95°C for 10 s, and annealing at 60°C for 40 s. The total volume was 20 μL (10 μL of 2×Premix Ex Taq,2 μL of cDNA, and 4 μL of each primer). Target geneexpression levels were normalized using β-actin, and expressed as the relative expression ratio of a target gene (relative mRNA value). All samples were run in triplicate and the results of qRT-PCR are shown as standard error (S.E.) of the means. The data were analyzed using SPSS 13.0 software. Treatment effects were examined by one-way analysis of variance(ANOVA) followed by Duncan’s multiple range test;results were considered statistically signi ficant when P <0.05.
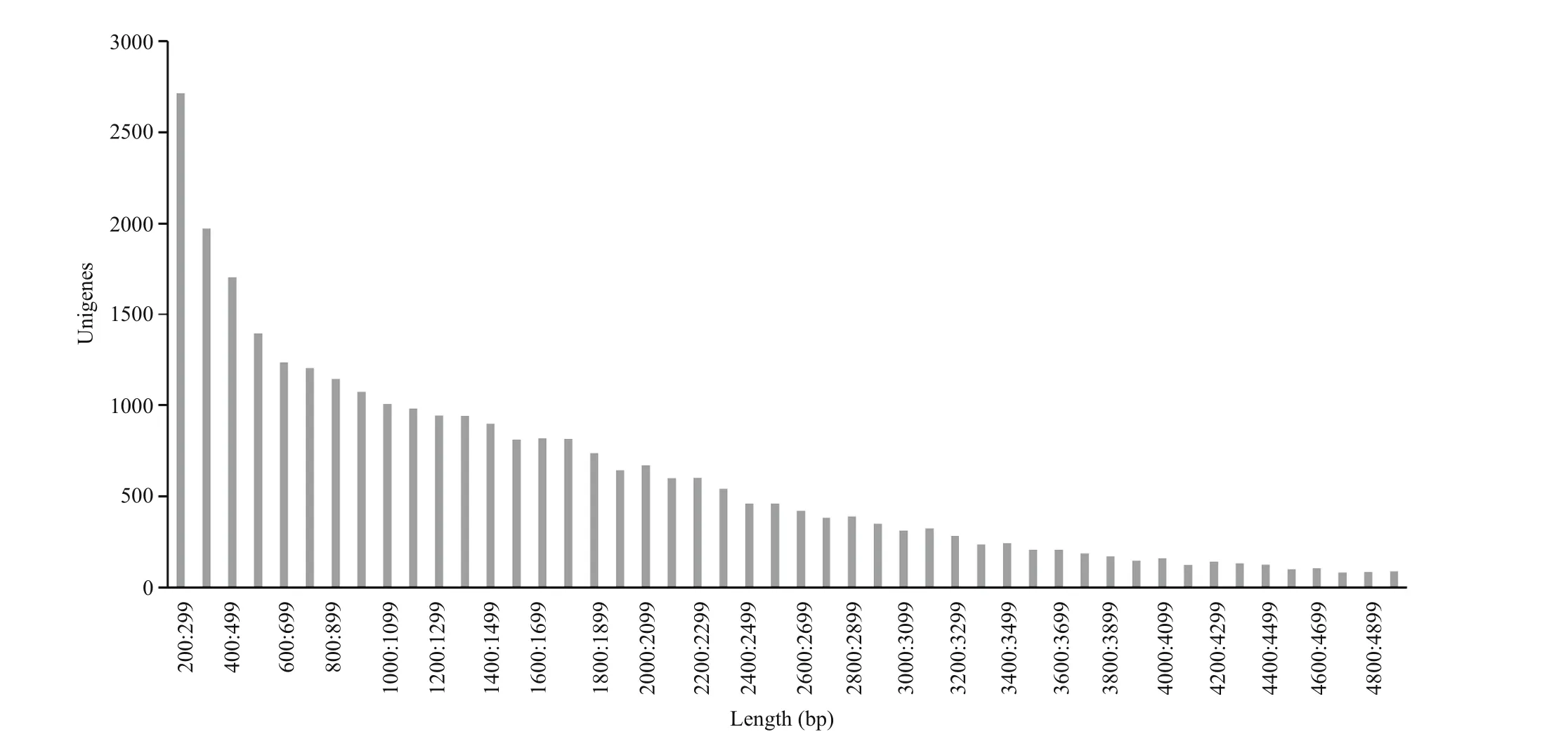
Fig.1 Length distribution of unigene of Pampus argenteus transcriptome
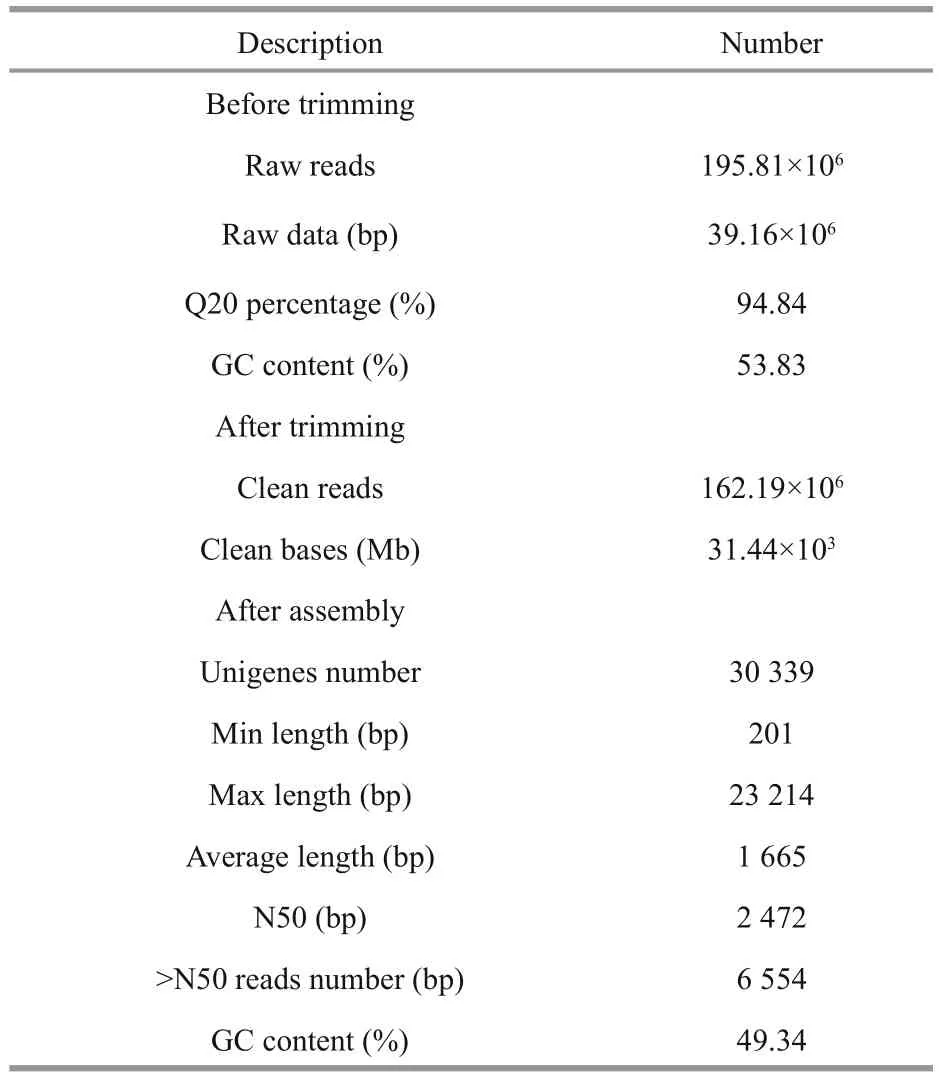
Table 2 Summary of sequences analysis
3 RESULT
3.1 Data filtering and de novo assembly
The Illumina sequencing data were deposited in the NCBI Database under accession number SAMN02440169. A summary of the analyzed sequences is shown in Table 2. In total, 195.81×106raw reads were generated. After filtering out lowquality reads, 162.19×106(82.8%) clean reads remained. Assembly of these reads resulted in 30 339 unigenes. The length of the obtained unigenes ranged from 201 to 23 214 bp. Length distributions of the Pampus argenteus unigenes are shown in Fig.1.
3.2 Annotation and functional assignment of the gene pathways, and global gene expression in silver pomfret
A total of 30 339 unigenes were annotated corresponding to unique protein accessions in the non-redundant protein database, and twenty-six clusters of eggNOG functional categories were de fined (Fig.2). GO analysis was conducted on those 30 339 unique proteins, using Blast2GO (http://www.BLAST2go.com) (Fig.3). According to the GO analysis, the majority of the genes had a molecular function, followed by genes assigned to a biological process and then to a cellular component. In brief, for the cellular component, genes involved in the cell(GO:0005623), cellular component (GO:0005575),intracellular component (GO:0005622), and organelles (GO:0043226) were highly represented;for biological process function, biological process(GO:0008150) was the most represented GO term,followed by the terms cellular nitrogen compound metabolic process (GO:034641), biosynthetic process(GO:0009058), and signal transduction(GO:0007165); the most represented categories for molecular function were molecular function(GO:0003674) and ion binding (GO:0043167).
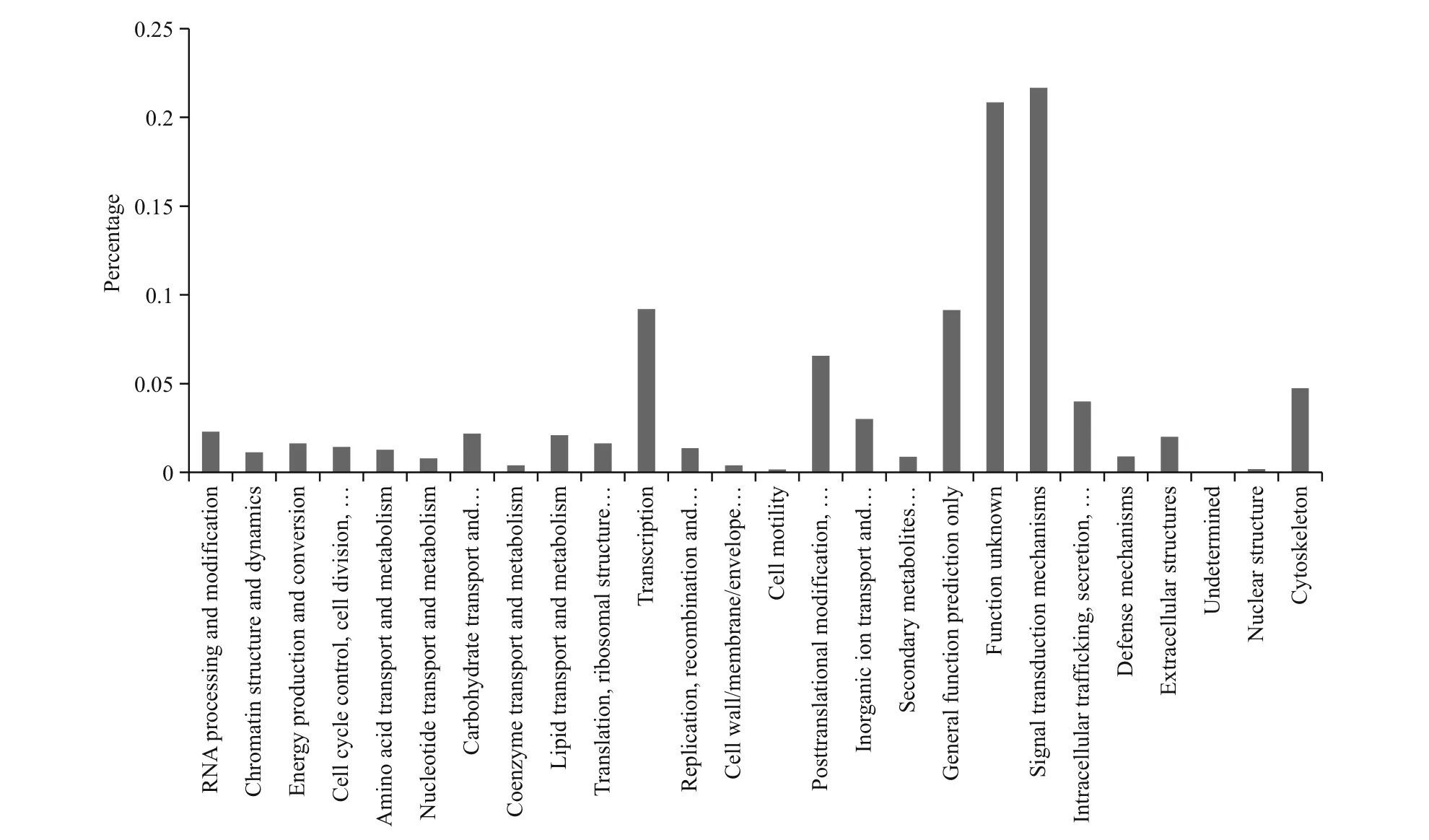
Fig.2 EggNOG functional category analysis in P. argenteus transcriptome
In addition, KEGG pathway analysis was performed using all unigenes as an alternative approach to functional categorization and annotation.Overall, 2 995 (9.87%) of the sequences were classi fied under terms for metabolism, including majority subgroups of carbohydrate metabolism(573), energy metabolism (262), lipid metabolism(509), nucleotide metabolism (312), amino-acid metabolism (408), and glycan biosynthesis and metabolism (460). And 1 530 sequences were grouped into a genetic information processing function,including transcription (214), translation (510),folding, sorting and degradation (544), and replication and repair (259). Environmental information processing, cellular processes, organismal systems,and human diseases groups, respectively, contained 2 289 (7.54%), 2 180 (7.19%), 5 083 (16.75%), and 6 056 (19.96%) KEGG annotated sequences.Moreover, KEGG pathway analysis was also performed based on separate unigenes in different response periods, and signi ficant differences were found between the periods of 24-h (response period)after stress (Fig.4a) and at 120-h (recovery period)after stress (Fig.4b). Compared with the response period, differentially expressed sequences that grouped into carbohydrate metabolism, lipid metabolism, xenobiotics biodegradation and metabolism, and endocrine system had decreased;however, genes related to amino-acid metabolism,biosynthesis of other secondary metabolism,metabolism of cofactors and vitamins, immune system, digestive system, nervous system,environmental adaptation, immune diseases,neurodegenerative diseases, and endocrine and metabolic diseases had increased during the recovery period. Examples of some differentially expressed genes are shown in Table 3.
3.3 Validation of transcriptomic data
RT-PCR was used to con firm the expression of genes that were identi fied in the Illumina sequencing analysis. The genes analyzed contained some metabolism, immune system and pathway-associated components, including the IGF/GH pathway,somatostatin-signaling pathway, and MAPK-signaling pathway. β-actin was used as an internal control to normalize the expression level. The results of RT-PCR showed different expression levels among the genes analyzed (Fig.5). The ppss 1 and cyp 21A genes showed the highest levels of expression,followed by cyp 11A, hsp 90A, ino 1 and tnfsf 13B; the fos gene displayed the lowest level of expression. The results were consistent with the results of transcriptome sequencing, and thus veri fied the accuracy and reliability of the protocol.
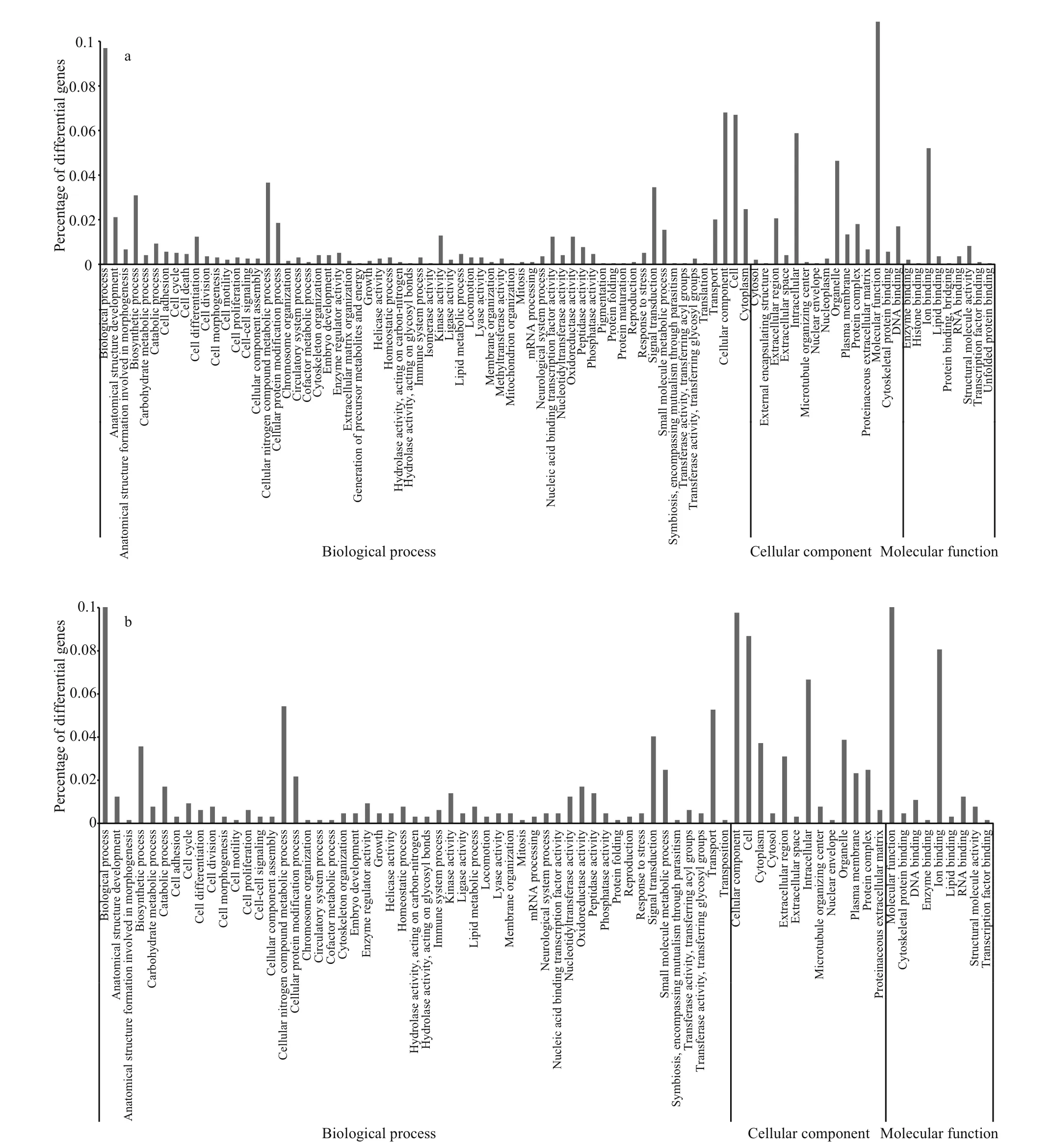
Fig.3 Gene ontology enrichment analysis at 24 h (a) and 120h (b) in P. argenteus
4 DISCUSSION
As a commercially important fish species, the silver pomfret Pampus argenteus has been studied since the 1980s (Almatar et al., 2004; Huang et al.,2010; Zhao et al., 2011; Sun et al., 2012, 2013; Liu et al., 2013). This species is sensitive to many types of stress, hence high mortality and low growth rate are often encountered in aquaculture endeavors. While stress factors greatly in fluence its production efficiency, the species is also useful as a representative specimen for studies of the stress response in marine fishes. To date, information about the genome and functional genes of Pampus species remains verylimited (Sun et al., 2017). In the present study, analysis of liver and kidney tissue transcriptome was conducted to generate a more comprehensive understanding of the silver pomfret genome, particularly changes in gene expression in response to handling stress. In a previous study, we divided the extended period following a stressor into a response period and a recovery period, based on fish behaviors and physiological parameters. In the same manner, the time points of 24 h and 120 h post-handling stress were chosen to determine the response status and the recovery status of P. argenteus in the present study.

Table 3 Differential expressed genes analysis in P. argenteus transcriptome
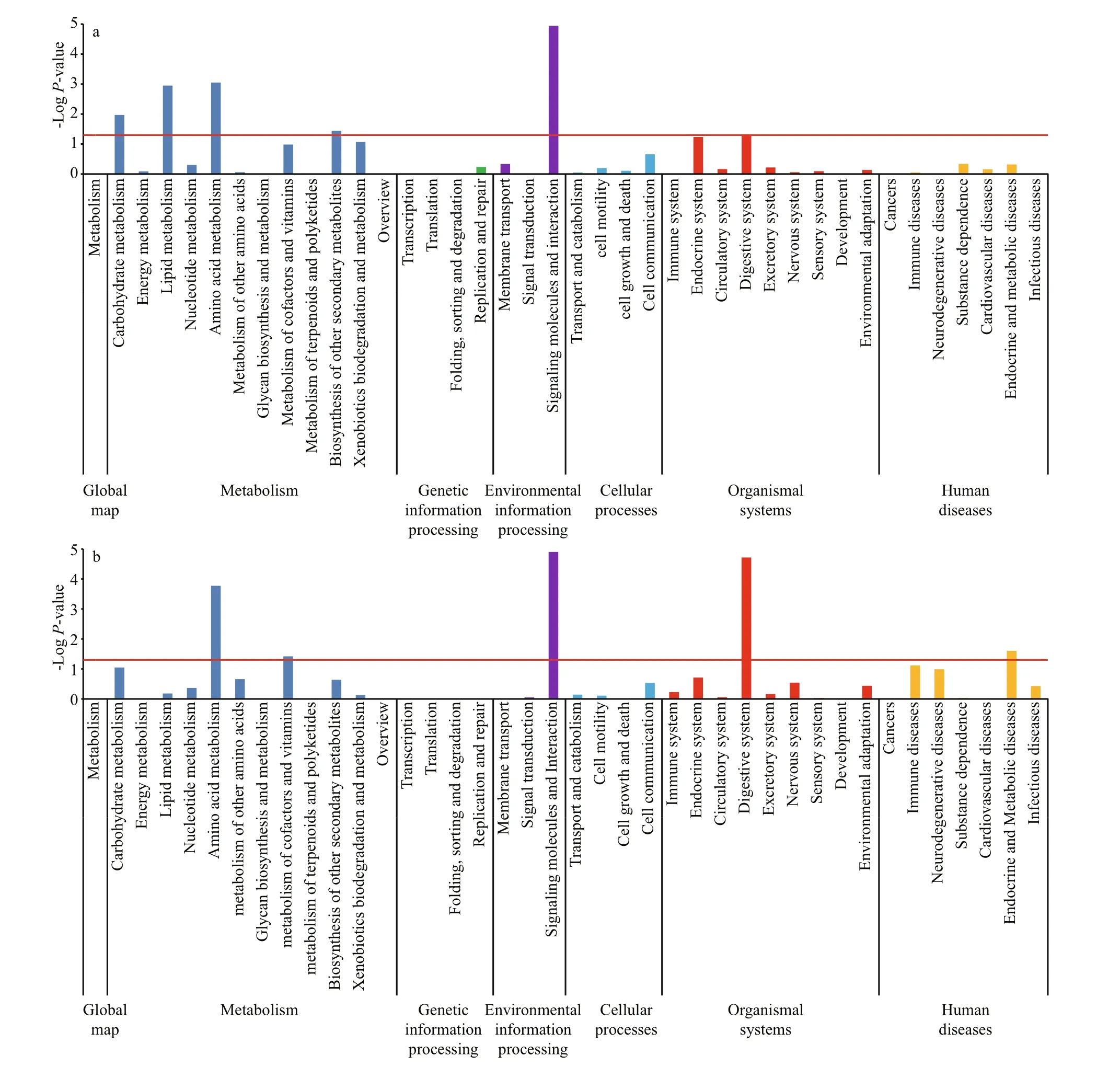
Fig.4 KEGG pathway enrichment analysis at 24 h (a) and 120 h (b) in P. argenteus
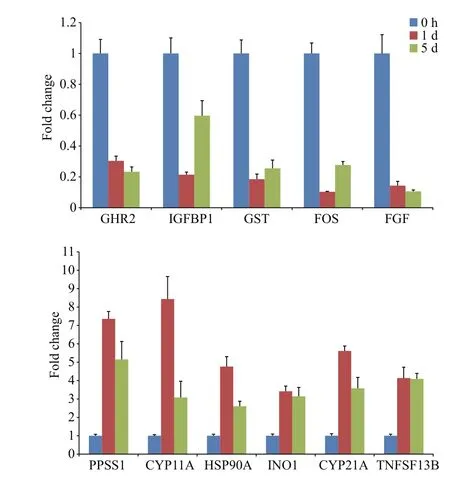
Fig.5 Quantitative real-time PCR con firmation of genes expressed in P. argenteus
The stress response in fish is a topic of much interest due to the impact of stressors on fish health and productivity in aquaculture systems, including adverse effects on growth, immunity, and even reproduction (Krasnov et al., 2005; Olsen et al., 2005;Hoskonen and Pirhonen, 2006; Momoda et al., 2007;Cairns et al., 2008; Bolasina, 2011). Our study aimed at assessing the time-course stress response in liver and kidney of P. argenteus, and identifying the occurrence of differentially expressed genes following handling stress in this commercially important species. According to GO and KEGG pathway analysis, different patterns of gene expression followed the handling-stress treatment. During the period of response (at 24 h), differentially expressed genes that had signi ficantly increased were mainly involved in the metabolic system (carbohydrate metabolism, lipid metabolism, amino-acid metabolism) and environmental informationprocessing system (signaling molecules and their interactions). In fish, the myo-inositol biosynthesis(MIB) pathway is reportedly important for myoinositol accumulation during response to a stressor.With the help of two key enzymes, namely myoinositol phosphate synthase (MIPS) and inositol monophosphatase (IMPA), the MIB pathway converts glucose-6-phosphate to myo-inositol, which protects cells from salinity stress (Geiger and Jin, 2006). Fiol et al. (2006) also revealed up-regulation of MIPS mRNA in response to acute hyperosmotic challenge in tilapia gill tissue. Likewise, signi ficant upregulation of MIPS mRNA in the present study also suggested a high energetic demand posed by handling stress in P. argenteus.
During the period of recovery (at 120 h), decreases occurred in differentially expressed gene sequences that were grouped into carbohydrate metabolism,lipid metabolism, xenobiotics biodegradation and metabolism, and endocrine system; whereas increases occurred in genes related to amino-acid metabolism,biosynthesis of other secondary metabolites,metabolism of cofactors and vitamins, immune system, digestive system, nervous system,environmental adaptation, immune diseases,neurodegenerative diseases, and endocrine and metabolic diseases. Nakano et al. (2013) reported on gene expression during the handling-stress response in coho salmon; their results showed that acute handling stress could down-regulate the expression of growth-related genes such as gh, ghr, and igf 1. In teleost fish, GHR2 are reported as functional receptors of GH (Kajimura et al., 2004; Pierce et al., 2005;Reindl et al., 2009). A trend of decreasing ghr 2 following stress treatment in P. argenteus indicated the adverse effect on growth in this species (Table 2).Heat-shock proteins (HSPs) are one group of proteins produced by cells in response to stress. Many members of this protein group act as chaperones to ensure correct protein folding or help to refold proteins damaged by stress. A high level of hsp 90 expression has been reported in many fish species subjected to various stressors, such as bacterial infections, heat stress, and food deprivation (Cara et al., 2005; Wu et al., 2012; Zhang et al., 2014; Xie et al., 2015; Zizza et al., 2017). In the present study, a high level of hsp 90A expression at 24 h following the handling-stress treatment indicated the important role played by HSP90A in damage repair incurred during the stress response (Table 3). Considering the decrease in its level of expression at 120 h during the stress response, we deduced a gradually impaired repair capability after long-term exposure to a stress condition in this fish. This conclusion was also consistent with expression trends among some of the immune-related genes. The complement system is known as an innate immune-defense mechanism for antigen recognition and effector functions. The activation of the complement system could result in assembly of complement complexes (such as complement component 9 [C9]), which characteristically induce complement-mediated cytolysis (Wang et al., 2013; Guo et al., 2016). Upregulation of C9 was also reported during a pathogen challenge in liver of rock bream Oplegnathus fasciatus(Wickramaarachchi et al., 2012). Interleukin-8 (IL-8)is a chemokine produced by macrophages and other cell types; it is also an important mediator of the immune reaction in the innate immune-system response (Heratha et al., 2016). In vertebrates, the major histocompatibility complex (MHC) had the function of pathogen resistance (Grimholt et al., 2003;Peters and Turner, 2008; Chen et al., 2017; Wu et al.,2017). During the period of recovery in the stress response, signi ficantly high levels of gene expression for C9, il-8, and MHC II mRNA were found at 120 h following the stress treatment in P. argenteus (Table 3). In the meantime, measures of the energy demand and sensitivity to pathogens were consistent with the species’ low growth rate and high mortality under culture conditions.
5 CONCLUSION
In this study, transcriptome pro filing was performed in the economically important fish species Pampus argenteus following handling stress. A total of 30 339 unigenes were annotated from undisturbed and stressed fish. The results showed that differentially expressed genes were mainly involved in pathways of metabolism, environmental information processing,organismal systems, and disease responses. In addition, according to GO and KEGG pathway analysis, differential expression patterns were found during the stress treatment. The differential expression patterns in the recovery and response periods suggested an adaptive nature in fish which allows for homeostatic recovery, but also suggests a maladaptive nature attended by negative effects on survival,growth, and immune response. The present study provides information that deepens our understanding of the genome and functional gene expression in a Pampus species. Furthermore, identi fication of genes involved in regulation of the stress response in a cultured fish could aid the measurement and monitoring of stressors under various conditions.
 Journal of Oceanology and Limnology2018年4期
Journal of Oceanology and Limnology2018年4期
- Journal of Oceanology and Limnology的其它文章
- Editorial Statement
- Effects of seawater acidi fication on the early development of sea urchin Glyptocidaris crenularis*
- Dietary effects of A zolla pinnata combined with exogenous digestive enzyme (Digestin™) on growth and nutrients utilization of freshwater prawn, Macrobrachium rosenbergii(de Man 1879)
- Preliminarily study on the maximum handling size, prey size and species selectivity of growth hormone transgenic and non-transgenic common carp Cyprinus carpio when foraging on gastropods*
- Hydrodynamic characteristics of the double-winged otter board in the deep waters of the Mauritanian Sea*
- De novo transcriptome sequencing reveals candidate genes involved in orange shell coloration of bay scallop Argopecten irradians*