
嵌合抗原受体T细胞治疗的研究进展
2018-07-26 15:55叶世光李萍梁爱斌
上海医药 2018年11期
叶世光 李萍 梁爱斌
摘 要 嵌合抗原受体(chimeric antigen receptor, CAR)是一种将抗体衍生的靶向片段与能激活细胞的信号传导结构域相连接而产生的重组免疫受体,可赋予T细胞不依赖于主要组织相容性复合体而直接识别肿瘤细胞表面抗原的能力。目前,临床研究已证实表达CAR的T细胞(CAR T cells, CAR-T)能有效地用于治疗难治或复发性的急性B淋巴细胞白血病,且由此促进了CAR-T用于治疗其他血液系统恶性肿瘤及实体瘤的研究。本文主要介绍CAR的基本设计,同时回顾CAR-T用于治疗B细胞白血病、淋巴瘤及数种实体瘤的研究结果,并讨论CAR-T治疗进一步发展所面临的主要挑战和如何提高其抗肿瘤效力问题。
关键词 嵌合抗原受体T细胞 血液系统恶性肿瘤 实体瘤
中图分类号:R730.51 文献标志码:A 文章编号:1006-1533(2018)11-0003-06
The current research of chimeric antigen receptor T cell therapy*
YE Shiguang, LI Ping, LIANG Aibin**
(Department of Hematology, Tongji Hospital, Tongji University, Shanghai 200065, China)
ABSTRACT Chimeric antigen receptor (CAR) is a recombinant immunoreceptor combining an antibody-derived targeting fragment with signaling domains capable of activating cells, which endows T cells with the ability of recognizing tumorassociated surface antigens independent of the expression of major histocompatibility complex. Recent early-phase clinical trials of CAR T cells (CAR-T) for relapsed or refractory B cell acute lymphoblastic leukemia have demonstrated promising results. Given this success, broadening the clinical experience of CAR-T therapy beyond hematological malignancies has been actively investigated. Here we discuss the basic design of CAR and review the clinical results from the studies of CAR-T therapy in B cell leukemia and lymphoma, and several solid tumors. We additionally discuss the major challenges in the further development and strategies for increasing anti-tumor activity and safety.
KEy WORDS chimeric antigen receptor T cell; hematological malignancies; solid tumors
嵌合抗原受体(chimeric antigen receptor, CAR)由细胞外抗原识别结构域(通常是抗体单链可变片段scFv)与细胞内信号传导结构域(T细胞受体的CD3 ζ链)相连接而成,其细胞外部分可使T细胞具有识别特异性抗原的能力。当CAR与其识别的抗原结合后便会通过信号传导结构域刺激T细胞增殖,同时激活T细胞的细胞毒作用并促进细胞因子分泌,最终消除带有该抗原的细胞。目前广泛使用的嵌合抗原受体T细胞(CAR T cells, CAR-T)治疗流程为:首先分离出患者自己的T细胞(或来自同种异体供者的T细胞),然后予以活化并进行基因修饰以获得CAR-T,最后回输至患者体内。此种治疗方法可极大地降低移植物抗宿主疾病风险,且可使脂类、蛋白质和碳水化合物这些抗原都能不受主要组织相容性复合体(major histocompatibility complex, MHC)的限制而被T细胞识别。另一种CAR-T治疗方法是设计出一种能够用于治疗所有表达相同抗原的肿瘤的CAR,这是一种更适于工业化生产、也便于临床应用的CAR-T治疗方法。
1 CAR的结构设计
CAR包含细胞外抗原识别结构域和细胞内信号传导结构域两个部分(图1)。其中,细胞外抗原识别结构域通常为scFv,后者通过铰链和(或)跨膜结构域锚定于细胞上,能够与肿瘤相关抗原(tumor-associated antigen, TAA)结合;细胞内信号传导结构域则是激活T细胞所必需的。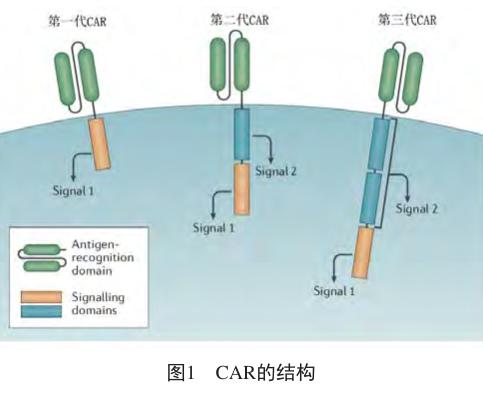
第一代CAR的细胞内信号传导结构域通常为T细胞受体的CD3 ζ链,可提供被称为“signal 1”的T细胞激活信号,但第一代CAR-T在临床试验中的表现不佳。尽管有证据表明,第一代靶向TAA的CAR-T可在人体内少量持续存在,然而其扩增能力和抗肿瘤效力很低[1]。
第二代CAR的细胞内信号传导结构域增加了一个共刺激信号传导结构域,故可同时提供signal 1和signal 2两种T细胞激活信号。与靶向CD19的第一代CAR-T(单独通过CD3 ζ链传导信号)相比,第二代CAR-T(同時通过CD3 ζ链和CD28传导信号)在被回输至非霍奇金淋巴瘤(non-Hodgkins lymphoma, NHL)患者体内后的存在时间更长、扩增能力更强。在过去的5年中,靶向CD19的第二代CAR-T[包含CD28或4-1BB(CD137)共刺激信号传导结构域]在治疗急性B淋巴细胞白血病(B-cell acute lymphocytic leukemia, B-ALL)方面显示具有显著的临床疗效[2],但有关第二代CAR的最佳细胞内信号传导结构域仍有待确定。
第三代CAR的细胞内信号传导结构域包含CD3 ζ链和2个共刺激信号传导结构域,共刺激信号由CD28、4-1BB或OX40(CD134)传导。临床前研究显示,与第二代CAR-T相比,第三代CAR-T表现出优异的抗肿瘤效力[3]。目前,一项临床试验(NCT01853631)正在招募患者,将比较第二代和第三代CAR-T治疗的抗肿瘤效果。这项研究将以NHL、B-ALL和慢性淋巴细胞白血病(chronic lymphocytic leukemia, CLL)患者为对象,分别给予靶向CD19的第二代CAR-T(通过CD3 ζ链和 CD28传导信号)和靶向CD19的第三代CAR-T(通过CD3 ζ链、4-1BB和CD28传导信号)治疗,研究结果可为未来CAR结构设计1个、还是2个共刺激信号传导结构域提供临床依据。
2 靶向CD19的CAR-T的临床研究
CAR-T治疗肿瘤的临床研究主要集中在CD19阳性的血液系统恶性肿瘤方面,包括B-ALL、CLL、滤泡性淋巴瘤(follicular lymphoma, FL)、弥漫性大B细胞淋巴瘤(diffuse large B-cell lymphoma, DLBCL)和套细胞淋巴瘤等。
2.1 治疗B-ALL
Kyrmiah(tisagenlecleucel)是诺华(Novartis)公司生产的CAR-T产品,2017年8月获美国FDA批准,用于治疗25岁以下的难治或复发性B-ALL患者。这是全球范围内首次批准一种CAR-T产品,同时也为肿瘤治疗开启了“以基因改造的免疫细胞治疗”的新治疗模式。Kyrmiah属第二代CAR-T(通过CD3 ζ链和4-1BB传导信号),最初由美国宾夕法尼亚大学开发,后于2012年8月将全球商业化开发权益转让给了诺华公司。Kyrmiah的关键临床试验是一项代号为“B2202”(NCT02435849)的Ⅱ期临床试验,后者为单组、多国、多中心臨床研究,研究地点包括美国、欧盟、加拿大、澳大利亚和日本等共11个国家。共有75例B-ALL患者接受了Kyrmiah治疗,结果显示3个月的总缓解率为81%(95% CI: 71% ~ 89%),6和12个月的无事件生存率和总生存率分别为73%(95% CI: 60% ~ 82%)、90%(95% CI: 81% ~ 95%)和50%(95% CI: 35% ~ 64%)、76%(95% CI: 63% ~ 86%)[4]。
2.2 治疗B细胞淋巴瘤
Yescarta(axicabtagene ciloleucel)是Kite制药公司生产的CAR-T产品,2017年10月获美国FDA批准,用于治疗经其他疗法治疗无效或既往至少接受过2种治疗方案治疗后又复发的特定类型成人大B细胞淋巴瘤,包括DLBCL、转化型FL和原发性纵隔B细胞淋巴瘤等患者。Yescarta是第一种被批准用于治疗某些特定类型的NHL的基因工程产品,属第二代CAR-T(通过CD3ζ链和CD28传导信号)。Yescarta的关键临床试验是一项代号为“ZUMA-1”的临床研究,其结果为Yescarta获得批准提供了安全性和有效性证据。该研究对101例接受Yescarta治疗的大B细胞淋巴瘤患者的临床数据进行了分析,发现总缓解率为82%,完全缓解率为54%。中位随访15.4个月,42%的患者有治疗反应,40%的患者持续完全缓解。18个月的总生存率为52%[5]。
3 用于血液系统恶性肿瘤治疗的CAR的新靶点
上述CAR-T产品都靶向CD19,故仅对CD19阳性的血液系统恶性肿瘤有效。不过,目前也在研究能够用于治疗更广泛血液系统恶性肿瘤的CAR的新靶点,其中最有希望的用于治疗B细胞肿瘤的CAR的新靶点包括CD22、CD20、无活性酪氨酸激酶跨膜受体ROR1和免疫球蛋白κ轻链(immunoglobulin κ, Igκ),用于治疗髓系肿瘤的CAR的新靶点包括CD123、CD33和LeY抗原,用于治疗多发性骨髓瘤(multiple myeloma, MM)的CAR的新靶点包括B细胞成熟抗原(B-cell maturation antigen, BCMA)和CD138。
3.1 用于治疗B细胞肿瘤的CAR的新靶点
CD22和CD20的表达模式与CD19类似,故靶向这些分子也可能导致非恶性B细胞的“靶向性、非肿瘤性”破坏,导致B细胞发育不良。这种毒性已见于靶向CD19的CAR-T治疗,但被认为是可接受的,因可通过每月经静脉输注免疫球蛋白来治疗[6]。目前,一项代号为“NCI34”的临床试验正在评估靶向CD22的CAR-T用于治疗FL、NHL、ALL和大B细胞淋巴瘤患者的临床效果。临床前研究结果表明,最佳抗CD22的CAR是第二代CAR,其scFv结合于近端CD22表位[7]。上述Ⅰ期临床试验(NCT02315612)将评估这种靶向CD22的CAR-T(通过CD3 ζ链和4-1BB传导信号)治疗CD22阳性的白血病或淋巴瘤患者的临床效果。
ROR1和Igκ在恶性B细胞上高表达,故这些蛋白可用作靶向B细胞肿瘤的CAR的有效靶点,且与靶向CD19、CD20或CD22的CAR相比,靶向ROR1或Igκ的CAR-T的“靶向性、非肿瘤性”毒性也可能较低。ROR1是一种糖基化的Ⅰ型膜蛋白,在许多肿瘤(包括CLL和B-ALL)细胞表面表达,但不表达于非恶性B细胞和造血干细胞。不过,有关ROR1的表达模式尚存争议,其可能在非肿瘤胰腺和肺组织中亦有表达[8]。如果这样,靶向ROR1的CAR-T的任何“靶向性、非肿瘤性”毒性都可能产生严重后果。未转化的B细胞表达Igκ或免疫球蛋白λ轻链,故靶向一种免疫球蛋白轻链的CAR-T对表达另一种免疫球蛋白轻链的B细胞没有毒性。与靶向Igκ有关的一个问题是,Igκ的可溶性形态似可通过与CAR结合并干扰其识别恶性B细胞上抗原的能力而降低CAR-T的抗肿瘤效力。好在临床前研究结果已经证实,靶向Igκ的CAR-T的抗肿瘤效力不受可溶性Igκ的影响。临床前研究结果表明,靶向ROR1的CAR可重定向T细胞功能,进而消除ROR1阳性的肿瘤细胞[9]。目前,一项Ⅰ期临床试验(NCT02194374)正在计划中,将评估靶向ROR1的CAR-T治疗CLL患者的临床效果。另一项Ⅰ期临床试验(NCT00881920)则在进行中,将评估靶向Igκ的CAR-T治疗B细胞白血病(包括CLL)、淋巴瘤和MM患者的临床效果。
CD30表达于Reed-Sternberg细胞上,这种细胞是霍奇金淋巴瘤(Hodgkins lymphoma, HL)患者特有的细胞类型,也是一小群活化的淋巴细胞。与靶向Igκ情况类似,靶向CD30的主要关注点也是经常发现患者有高水平的可溶性CD30。这些可溶性CD30会与CAR结合,导致CAR与肿瘤细胞表面的CD30结合的能力减弱。不过,临床前研究结果表明,可溶性CD30亦不会影响靶向CD30的CAR-T的抗肿瘤效力[10]。目前,一项临床研究(NCT01316146)正在计划中,将评估靶向CD30的CAR-T治疗HL和NHL患者的临床效果。
3.2 用于治疗髓系肿瘤的CAR的新靶点
用于治疗髓系肿瘤的CAR的新靶点主要包括CD123、CD33和LeY抗原。鉴于这些分子也可在一些非恶性细胞上表达,靶向这些抗原的CAR-T可能存在一定的“靶向性、非肿瘤性”毒性。尽管临床试验已证实靶向CD123的单克隆抗体治疗安全[11],但因CAR-T的作用模式不同,故仍需小心评估靶向这些抗原的CAR-T治疗的安全性。
CD123表达于恶性髓系细胞上,但也表达于内皮细胞和单核细胞上,因此靶向CD123的CAR-T有一些“靶向性、非肿瘤性”毒性[12]。目前,一项临床试验(NCT02159495)正在评估靶向CD123的CAR-T治疗急性髓细胞白血病(acute myeloid leukemia, AML)患者的安全性及有效性。
CD33表达于恶性髓系细胞上,但靶向该蛋白的CAR-T可能存在一些问题,因为一些T细胞和造血干细胞也表达CD33,故可能导致患者出现长时间的血细胞计数减少和造血功能恢复延迟。此外,靶向CD33的CAR-T也可能存在肝毒性,因为CD33也表达于肝脏的库普夫细胞上[13]。国内目前正在进行一项临床试验(NCT01864902),将评估靶向CD33的CAR-T治疗AML患者的安全性及有效性。据此试验的病例报告,治疗的毒性反应包括发热和持续的全血细胞计数减少,提示靶向CD33的CAR-T有用于造血干细胞移植前清髓的潜力[14]。
LeY抗原是一种表达于部分AML细胞上的岩藻糖基化的糖类抗原,但也表达于胃肠和胰腺细胞上[15]。一项临床试验(NCT01716364)使用靶向LeY抗原的CAR-T治疗4例AML患者,结果在1例患者中观察到CAR-T能够到达骨髓并介导一些抗肿瘤免疫反应,但所有患者最终都出现了复发[16]。使用靶向LeY抗原的CAR-T治疗AML似是安全的,但单以靶向LeY抗原的CAR-T治疗AML的临床效果有限,可能与LeY抗原仅表达于部分AML细胞上有关。
3.3 用于治疗MM的CAR的新靶点
用于治疗MM的CAR的新靶点包括CD138、BCMA和CD19。CD138是一种Ⅰ型整合膜硫酸乙酰肝素蛋白多糖,不仅表达于浆细胞和MM细胞上,也表达于支气管上皮上,故肺毒性可能是靶向CD138的CAR-T的主要毒性[17]。一项临床试验(NCT01886976)正在评估靶向CD138的CAR-T治疗MM患者的临床效果。初步研究结果显示,5例患者中有4例达到疾病稳定,3级不良反应仅为发热,没有剂量限制性毒性,治疗的可耐受性良好[18]。
BCMA属肿瘤坏死因子超家族的细胞表面受体,仅表达于浆细胞上。因此,使用靶向BCMA的CAR-T治疗可能会导致根除人体内的浆细胞,包括恶性和非恶性浆细胞,但可经静脉输注免疫球蛋白有效控制这种“靶向性、非肿瘤性”毒性。表达模式和临床前研究结果均表明,BCMA是CAR-T治疗MM的一个很有吸引力的靶分子[19]。目前,一项临床试验(NCT02215967)正在进行中,将评估靶向BCMA的CAR-T治疗MM或浆细胞骨髓瘤患者的临床效果。
4 用于实体瘤治疗的CAR的靶点
人们也在开发用于治疗实体瘤的CAR-T,并为此鉴定了一些CAR的新靶点。
前列腺特异性膜抗原(prostate-specific membrane antigen, PSMA)是一种Ⅱ型膜蛋白,在大多数前列腺癌细胞上以及许多实体瘤相关的新生血管系统中呈高表达状态[20]。因此,靶向PSMA的CAR-T可能具有抗新生血管作用,甚至具有直接的抗肿瘤活性。目前,一项临床试验(NCT01140373)正在评估靶向PSMA的第二代CAR-T治疗前列腺癌的临床效果。
间皮素是一种表达于恶性胸膜间皮瘤以及卵巢癌、胰腺癌和一些肺癌细胞上的TAA,同时也表达于正常的腹膜、胸膜和心包间皮细胞上。目前,一项临床试验(NCT02159716)正在评估靶向间皮素并用慢病毒转导的CAR-T(以实现稳定的CAR表达)治疗转移性胰腺导管腺癌、上皮性卵巢癌和恶性上皮性胸膜间皮瘤患者的临床效果。另一项临床试验(NCT02414269)则将评估另一种靶向间皮素的第二代CAR-T的临床效果,现已有一些罹患恶性胸膜疾病、间皮瘤、乳腺癌或间皮素阳性的肺癌患者入选该研究,他们将接受经腹膜输注此CAR-T的治疗。
现也在评估靶向表皮生长因子受体(epidermal growth factor receptor, EGFR)和EGFR變异体的CAR-T治疗胶质瘤患者的临床效果。EGFR在许多类型的肿瘤中过度表达,并与肿瘤侵袭和转移相关。EGFR和EGFR变异体Ⅲ的突变形态也常常在肿瘤中过度表达,且通常与胶质母细胞瘤相关。临床研究已经证实,靶向EGFR变异体的单克隆抗体治疗的耐受性良好[21],提示EGFR变异体Ⅲ可能也是CAR-T治疗的适宜靶点。目前,一项临床试验(NCT02209376)正在招募难治或复发性胶质瘤患者,将评估靶向EGFR变异体Ⅲ的CAR-T治疗的安全性和有效性。
5 提高CAR-T抗肿瘤效力的新策略
由于现有CAR仍有许多不足,故人们也在研究一些新的策略,以期能够提高CAR-T治疗的抗肿瘤效力。
5.1 “Armoured”CAR
“Armoured”CAR是经进一步修饰的CAR,它们能够分泌促炎性细胞因子来保护自身免受抑制性肿瘤微环境的影响。临床前研究表明,与常规CAR-T相比,经修饰能分泌白介素-12的“Armoured”CAR-T具有更高的抗肿瘤效力[22]。目前,一项临床试验(NCT02498912)正在招募黏蛋白-16(在卵巢癌等细胞上高表达)阳性的复发性卵巢癌、输卵管癌和原发性腹膜癌患者,将评估由第二代CAR与黏蛋白-16的胞外域结合而成、由此靶向黏蛋白-16并能分泌白介素-12的CAR-T治疗的安全性和有效性。
5.2 双受体或趋化因子受体CAR
为了改善CAR-T在人体内的持久性,研究人员设计了同时表达两种受体的CAR-T,一种用于TAA的识别,另一种则为T细胞提供增殖刺激信号。例如,先将第二代CAR的信号传导结构域与TAA识别受体T1E28z相连接,然后再将此CAR与由人表皮生长因子受体(human epidermal growth factor receptor, HER)1的同源二聚体和HER2-HER3异源二聚体结合形成的多肽相连接[23]。趋化因子受体CAR由CAR与白介素-4受体结合而成,现正在进行用于治疗头颈部肿瘤患者的临床试验(NCT01818323)。
5.3 自然杀伤细胞受体CAR
自然杀伤细胞会表达能够分辨恶性和非恶性细胞的受体,后者也可被用作CAR的抗原识别结构域以提高T细胞对肿瘤细胞的识别能力,其中一种这样的受体就是NKG2-D。临床前研究结果证实,将NKG2-D的胞外结构域连接到CAR的信号传导结构域后能够激活T细胞并产生抗肿瘤效力[24]。目前,一项临床研究正在评估这种CAR-T治疗的安全性和抗肿瘤效力。
5.4 联用细胞周期阻断剂
CAR-T很容易受到肿瘤微环境中存在的抑制性免疫检查点信号如程序性细胞死亡蛋白-1(programmed cell death protein-1, PD-1)配体和细胞毒性T淋巴细胞相关抗原-4(cytotoxic T lymphocyte-associated antigen-4, CTLA-4)配体等的影响。已有研究发现,使用这些配体的抗体可恢复内源性肿瘤特异性T细胞反应。事实上,CAR-T对PD-1介导的免疫抑制也是敏感的[25]。因此,CAR-T与免疫检查点抑制信号的单克隆抗体联用能够保护CAR-T的抗肿瘤效力免受肿瘤微环境的影响。目前,一项临床试验(NCT00586391)正在评估靶向CD19的CAR-T联合ipilumimab(一种CTLA-4阻断性单克隆抗体)治疗B细胞淋巴瘤、CLL和B-ALL患者的临床效果。
5.5 延长CAR-T的存在时间
CAR-T治疗失败的一个潜在原因是由于人体内的免疫系统识别出CAR为外来肽,从而引起免疫介导的CAR-T的破坏。克服这种免疫介导破坏的一种方法是清除B细胞,以防止宿主产生CAR特异性的人抗小鼠肽抗体,后者会导致CAR-T的消除[26]。目前,一项临床试验(NCT02465983)正在评估靶向间皮素的CAR-T联合靶向CD19的CAR-T治疗胰腺癌患者的临床效果。
5.6 靶向肿瘤血管系统的CAR-T
肿瘤微环境中血管内皮生长因子(vascular endothelial growth factor, VEGF)的存在和肿瘤细胞上VEGF受体的过度表達均与肿瘤转移及其患者的不良预后有关。VEGF受体-2在肿瘤基质细胞和某些肿瘤细胞上呈过度表达状态[27]。临床前研究结果表明,靶向VEGF受体-2的CAR可将T细胞功能重定向至肿瘤基质细胞,同时保留正常的基质细胞。目前,一项临床试验(NCT01218867)正在评估靶向VEGF受体-2的CAR-T治疗转移性黑素瘤和肾癌患者的临床效果。
6 结语
CAR-T已被证实是难治和复发性B细胞肿瘤的一种有效治疗手段。在CAR-T治疗难治和复发性B-ALL及B细胞淋巴瘤取得成功之后,人们开始设计各种不同的CAR-T,以期能够用于有效治疗其他血液系统恶性肿瘤和实体瘤,但面临对目标抗原的谨慎选择、“靶向性、非肿瘤性”毒性的管理和对免疫抑制性肿瘤微环境的调控等问题的挑战。目前正在进行的各项临床试验的结果令人期待,希望它们能够为开发更多的更安全、更有效的CAR-T提供指导。
参考文献
[1] Jensen MC, Popplewell L, Cooper LJ, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans [J]. Biol Blood Marrow Transplant, 2010, 16(9): 1245-1256.
[2] Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia [J]. N Engl J Med, 2014, 371(16): 1507-1517.
[3] Zhong XS, Matsushita M, Plotkin J, et al. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cellmediated tumor eradication [J]. Mol Ther, 2010, 18(2): 413-420.
[4] Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia[J]. N Engl J Med, 2018, 378(5): 439-448.
[5] Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-Cell therapy in refractory large B-cell lymphoma [J]. N Engl J Med, 2017, 377(26): 2531-2544.
[6] Dotti G, Gottschalk S, Savoldo B, et al. Design and development of therapies using chimeric antigen receptorexpressing T cells [J]. Immunol Rev, 2014, 257(1): 107-126.
[7] Haso W, Lee DW, Shah NN, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia [J]. Blood, 2013, 121(7): 1165-1174.
[8] Hudecek M, Schmitt TM, Baskar S, et al. The B-cell tumorassociated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor [J]. Blood, 2010, 116(22): 4532-4541.
[9] Vera J, Savoldo B, Vigouroux S, et al. T lymphocytes redirected against the κ light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells[J]. Blood, 2006, 108(12): 3890-3897.
[10] Hombach A, Heuser C, Sircar R, et al. An anti-CD30 chimeric receptor that mediates CD3-ζ-independent T-cell activation against Hodgkins lymphoma cells in the presence of soluble CD30 [J]. Cancer Res, 1998, 58(6): 1116-1119.
[11] Tettamanti S, Marin V, Pizzitola I, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor [J]. Br J Haematol, 2013, 161(3): 389-401.
[12] Mardiros A, Dos Santos C, McDonald T, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia [J]. Blood, 2013, 122(18): 3138-3148.
[13] Sievers EL, Larson RA, Stadtmauer EA, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse [J]. J Clin Oncol, 2001, 19(13): 3244-3254.
[14] Wang QS, Wang Y, Lv HY, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia [J]. Mol Ther, 2015, 23(1): 184-191.
[15] Peinert S, Prince HM, Guru PM, et al. Gene-modified T cells as immunotherapy for multiple myeloma and acute myeloid leukemia expressing the Lewis Y antigen [J]. Gene Ther, 2010, 17(5): 678-686.
[16] Ritchie DS, Neeson PJ, Khot A, et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia [J]. Mol Ther, 2013, 21(11): 2122-2129.
[17] Zhang H, Xia T, Meng H, et al. Differential expression of syndecan-1 mediates cationic nanoparticle toxicity in undifferentiated versus differentiated normal human bronchial epithelial cells [J]. ACS Nano, 2011, 5(4): 2756-2769.
[18] Guo B, Chen MX, Han QW, et al. CD138-directed adoptive immunotherapy of chimeric antigen receptor (CAR)-modified T cells for multiple myeloma [J]. J Cell Immunother, 2016, 2(1): 28-35.
[19] Carpenter RO, Evbuomwan MO, Pittaluga S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma [J]. Clin Cancer Res, 2013, 19(8): 2048-2060.
[20] Haffner MC, Kronberger IE, Ross JS, et al. Prostate-specific membrane antigen expression in the neovasculature of gastric and colorectal cancers [J]. Hum Pathol, 2009, 40(12): 1754-1761.
[21] Thomas M. Cetuximab: adverse event profile and recommendations for toxicity management [J]. Clin J Oncol Nurs, 2005, 9(3): 332-338.
[22] Pegram HJ, Lee JC, Hayman EG, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning [J]. Blood, 2012, 119(18): 4133-4141.
[23] Davies DM, Foster J, Van Der Stegen SJ, et al. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells [J/OL]. Mol Med, 2012, 18: 565-576 [2018-01-21]. doi: 10.2119/molmed.2011.00493.
[24] Spear P, Barber A, Rynda-Apple A, et al. NKG2D CAR T-cell therapy inhibits the growth of NKG2D ligand heterogeneous tumors [J]. Immunol Cell Biol, 2013, 91(6): 435-440.
[25] Choudhury N, Nakamura Y. Importance of immunopharmacogenomics in cancer treatment: patient selection and monitoring for immune checkpoint antibodies [J]. Cancer Sci, 2016, 107(2): 107-115.
[26] Kershaw MH, Westwood JA, Parker LL, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer [J]. Clin Cancer Res, 2006, 12(20 Pt 1): 6106-6115.
[27] Chinnasamy D, Yu Z, Theoret MR, et al. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice [J]. J Clin Invest, 2010, 120(11): 3953-3968.
