
分散固相萃取结合HPLC-MS/MS同时测定茶叶中丁醚脲及其降解产物
2018-07-14 03:15:38肖利民王美玲戴洁芸
食品与机械 2018年5期
肖利民 王美玲 戴洁芸 戴 华
(1. 长沙理工大学化学与生物工程学院,湖南 长沙 410076;2. 湖南出入境检验检疫局,湖南 长沙 410004)
茶叶是中国重要的农产品之一,在生产过程中,为了保证茶叶的产量,防治茶树的病虫害,农药的用量和种类也逐渐增多,使得农药残留成为茶叶安全最突出的问题之一。丁醚脲是一种新型高效的硫脲类选择性杀虫、杀螨剂,具有较低毒性,主要通过影响昆虫呼吸作用、干扰能量转换和转化为具有更高毒性的降解产物,达到杀虫目的[1],被广泛用于茶树上,对蚜虫、叶蝉、粉虱、蛾和螨类具有很好的杀灭效果[2-3]。但丁醚脲易发生光解、水解以及生物降解,也可在植物体内发生降解,形成丁醚脲-酰胺体、丁醚脲-脲等降解产物[4]。研究结果[5]表明丁醚脲的降解产物,比丁醚脲原药本身具有更高的毒性。这些分解产物还对肺、肝脏等靶器官的毒性具有积累作用[6]。随着丁醚脲的广泛使用,必然会对环境及食物造成污染,威胁人类健康。为此一些发达国家和地区都制定了严格的茶叶中丁醚脲残留限量标准。欧盟、美国等规定不得检出,欧盟于2002 年颁布了第2076/2002 号法规,明令禁止使用和销售丁醚脲。日本、澳大利亚等国家规定茶叶中丁醚脲及其2种降解产物的最大残留限量总值为20 mg/kg。中国GB 2763—2016《食品安全国家标准 食品中农药最大残留限量》规定了丁醚脲在茶叶中临时最大残留限量为5 mg/kg。与欧盟标准相比,中国规定的茶叶中丁醚脲的残留限量较高,同时未说明应包括丁醚脲降解产物包含在内,造成丁醚脲残留检测结果偏低或检测不出,给茶叶中丁醚脲农药残留的控制带来很大影响,也达不到出口茶叶丁醚脲残留限量检测要求。因此,建立茶叶中丁醚脲及其降解产物残留量的检测方法具有重要意义。
目前报道的主要是水[7]、蔬菜[8]、水果及土壤等[9-10]基质中丁醚脲残留分析的方法,虽有少数文献报道了茶叶中丁醚脲残留的检测,如吕慧芝等[11]采用QuEChERS-高效液相色谱液质联用法测定茶叶中的丁醚脲残留量,定量限为0.01 mg/kg,回收率为70.4%~110.1%,但只包括丁醚脲原药降解产物分析;董晓倩等[12]建立的茶叶中丁醚脲农药残留检测的液相色谱—串联质谱方法,研究了丁醚脲在紫外光下的分解,及丁醚脲—甲酰胺、丁醚脲—脲的质谱检测方法,没有定量检测数据;张新忠等[13]采用超高效液相色谱—串联质谱建立了测定茶叶和土壤中丁醚脲及其1种降解产物——丁醚脲—脲残留量的方法。由于丁醚脲的半衰期较短,茶叶中的丁醚脲在生产、运输、存储过程以及样品检测分析过程中都会分解、降解,产生丁醚脲—甲酰胺、丁醚脲—脲等降解产物,大多情况下,因为丁醚脲的降解,导致样品中难以检测到丁醚脲原药的残留,因此茶叶中丁醚脲农药的残留量应以丁醚脲及其降解产物总量来计算。不对茶叶中丁醚脲及其2种降解产物进行同时定性、定量分析,易造成漏检、检测结果偏低,不能准确反映茶叶中丁醚脲的实际残留量,达不到残留水平监测的目的。
茶叶样品基体复杂,杂质干扰严重,同时因为丁醚脲的降解、异构化、衍生作用,给分离检测技术带来很大的阻碍。本研究拟采用高效液相色谱—串联质谱技术,结合QuEChERS净化方法,建立茶叶中丁醚脲及其2种降解产物(丁醚脲—脲及丁醚脲—甲酰胺)残留量同时定性、定量测定的方法。
1 材料与方法
1.1 仪器与材料
三重四级杆串联质谱仪:API 4000型,配电喷雾离子源(ESI),美国Applied Biosystems公司;
高效液相色谱仪:SHIMADZU LC-20A型,日本岛津公司;
超纯水系统:MILLI-Q Gradient型,美国密理博公司;
快速混匀器:SK-1型,常州澳华仪器有限公司;
切割式混合研磨仪:GM200型,德国Retsch公司;
高速离心机:1-15P型,德国Sigma公司;
丁醚脲标准品:纯度>99.9%,德国Dr. Ehrenstorfer GmbH公司;
丁醚脲—脲、丁醚脲—甲酰胺标准品:纯度>99.0%,日本和光纯药工业株式会社;
甲醇、乙腈、乙酸:色谱纯,德国Merck公司;
其他试剂均为分析纯;
试验用水:超纯水,美国Millipore超纯水仪制备;
N-丙基乙二胺(PSA)吸附剂(40~60 mm)、石墨化炭黑(GCB)吸附剂(120~400目):上海安谱科学仪器有限公司;
茶叶:市售。
1.2 标准溶液配制
分别称取10.0 mg丁醚脲、丁醚脲—脲和丁醚脲—甲酰胺标准品,置于10 mL棕色容量瓶中,用乙腈溶解并定容配制成1.0 mg/mL的标准储备液,并转移至棕色样品瓶中,-20 ℃ 避光下保存以防止丁醚脲分解。将标准贮备溶液用空白基质逐级稀释为标准工作溶液,现用现配。
1.3 样品前处理
1.3.1 提取 称取粉碎后的茶叶样品1 g(精确至0.001 g)于50 mL塑料离心管中(铝箔纸包裹),加入0.2 g无水硫酸镁、0.2 g氯化钠和10 mL乙腈,涡旋混匀3 min,振荡提取10 min,于6 000 r/min离心3 min,收集上清液,待净化。
1.3.2 净化 取上清液1.0 mL于1.5 mL离心管中,加入0.15 g 无水硫酸镁、0.05 g GCB吸附剂和0.05 g PSA吸附剂,旋涡振荡1 min,10 000 r/min离心3 min。取净化液经0.22 μm有机微孔滤膜过滤于棕色进样小瓶,待测定。
1.4 色谱条件
色谱柱:Agilent ZORBAX Extend-C18柱(150 mm×4.6 mm,3.5 μm);流动相A为0. 1%乙酸水溶液,流动相B为乙腈;梯度洗脱程序:0~4 min、20% B~70% B,4~8 min、70% B~95% B,8~12 min、95% B,12.0~12.1 min、95% B~20% B,12.1~18.0 min、20% B;柱温:40 ℃,进样量:10 mL。
1.5 质谱条件
离子源:ESI,正离子扫描;气帘气:68.9 kPa;雾化气 (GS1):275.8 kPa;辅助加热气 (GS2):344.8 kPa;碰撞气 (CAD):48.3 kPa;电喷雾电压 (IS):5 000 V;离子源温度 (TEM):450 ℃;定性离子对、定量离子对、去簇电压(DP)和碰撞电压(CE),具体见表1。
2 结果与讨论
2.1 色谱和质谱条件的优化
分别考察了甲醇—水、乙腈—水(含0.1%乙酸)以及乙腈—5 mmol/L 乙酸铵3种流动相体系对分离效果及灵敏度等影响。结果发现,以甲醇—水或乙腈—5 mmol/L乙酸铵作为流动相时,丁醚脲峰型较宽,响应不高。采用乙腈—0.1% 乙酸作为流动相以及梯度洗脱模式,3种化合物均可获得较好的分离,峰形尖锐且响应值高,杂质干扰少,故确定为最终的液相条件。在此色谱条件下,出峰时间顺序依次为丁醚脲—甲酰胺、丁醚脲—脲和丁醚脲。
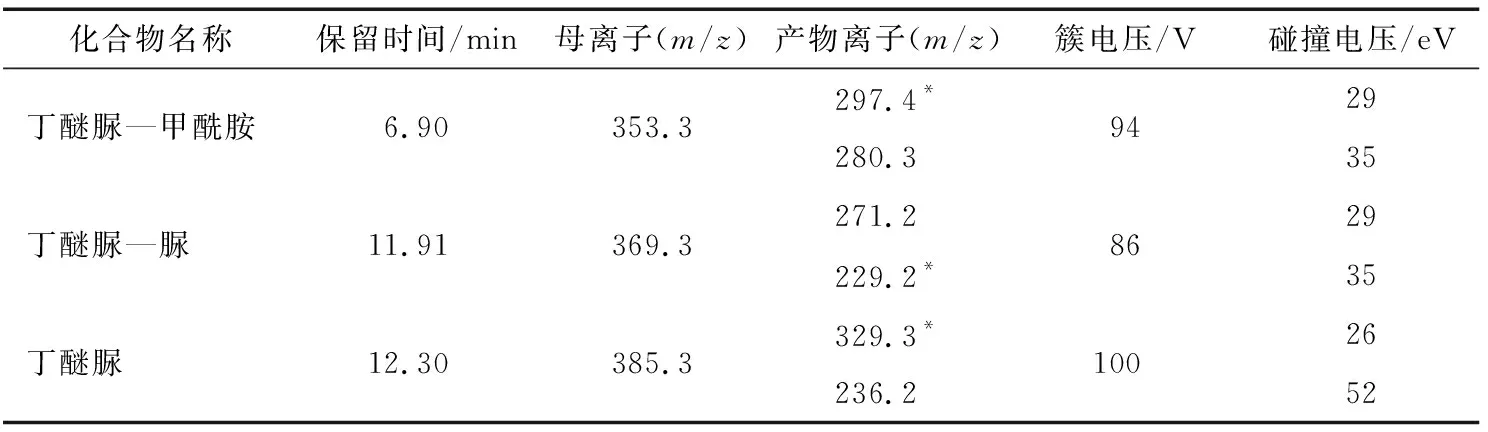
表1 丁醚脲及其降解产物的质谱参数†Table1 HPLC-MS/MS parameters for diafenthiuron and its degradation products
† “*”表示定量离子对。
丁醚脲及其降解物在ESI+或ESI-离子模式下均有响应,因该类化合物均含有氨基基团,易在正模式下形成[M+H]+离子,因而选取ESI+模式测定,灵敏度更高。同时对质谱的离子源温度、雾化气、辅助加热气等参数进行优化,使各化合物的质谱响应信号达到最佳。
2.2 样品提取和净化条件优化
考察了乙腈、甲醇、丙酮、乙酸乙酯和二氯甲烷等有机溶剂对目标化合物提取效率的影响。结果表明,相对于其他溶剂,采用乙腈作为提取剂,因其渗透性强、溶解性良好,不仅可获得较高的提取回收率,而且提取液中茶多酚、咖啡碱、色素等共萃物较少,有利于后续的进一步净化[12-13]。试验中比较了加水浸泡与不加水浸泡对于干茶叶中丁醚脲及其降解产物提取的影响。结果表明,加水浸泡反而降低干茶叶样品中丁醚脲及其降解产物的提取率。一方面是丁醚脲含有硫脲基在见光及水环境中易降解,图1为丁醚脲在水溶液(200 ng/mL)中室温放置24 h后的降解情况。由图1可见,丁醚脲在水溶液中分解速度较快,0.5 h后便降解64%,同时检测到2种降解产物丁醚脲—甲酰胺和丁醚脲—脲。丁醚脲—甲酰胺的浓度随时间不断增加,24 h后浓度增加26倍。而丁醚脲—脲自前0.5 h浓度增加为原来的2.5倍,之后随时间的延长浓度基本不变。这表明丁醚脲在见光及水环境中易降解,但除了降解产生丁醚脲—甲酰胺和丁醚脲—脲外(图2),还可能有其他机理的降解发生[6],需进一步研究。另一方面,加水浸泡后,提取液色素明显加重,一些极性物质会被提取出来,干扰更严重。茶叶中富含大量生物碱、色素和多酚类物质,基质干扰严重,一般需采用固相萃取柱或凝胶渗透色谱净化,再经过浓缩、复溶等过程。不仅前处理时间较长,步骤繁琐,而且由于丁醚脲的不稳定性,还会导致丁醚脲的分解,使得回收率偏低。因此简单、快速的前处理净化方法有助于减少或避免丁醚脲的分解,准确地获得样品中丁醚脲及其降解产物的实际残留量。本研究参考已有文献[11-12]方法,采用QuEChERS净化法, PSA吸附剂用于去除茶叶提取液中的脂肪酸、酚类等杂质,GCB吸附除茶叶提取液中色素成分。无水硫酸镁去除提取液中的水分,提高回收率。该方法简单、快速、前处理用时短。由于丁醚脲见光容易降解,因此试验过程中要注意避光,采用的塑料离心管需用铝箔纸包裹。
2.3 基质效应
液相色谱-质谱进行检测时,由于基质成分和目标化合物在离子化时存在相互竞争,常常会增强或抑制待测物的离子化作用,从而影响分析结果的准确性。本研究分别以乙腈和空白茶叶样品基质配制混合标准工作溶液,以峰面积比为纵坐标、质量浓度为横坐标绘制标准曲线(图3),比较两者的斜率以确定基质效应的强弱。试验结果发现,不同茶叶样品中丁醚脲和丁醚脲—脲均存在一定的基质抑制效应。因此采用空白样品基质溶液配制标准工作溶液进行定量分析,以消除基质效应的影响。
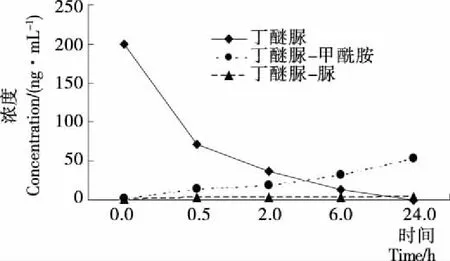
图1 在水溶液中丁醚脲(200 ng/mL)随时间的变化
Figure 1 The changes of diafenthiuron in aqueous solution at the concentration of 200 ng/mL
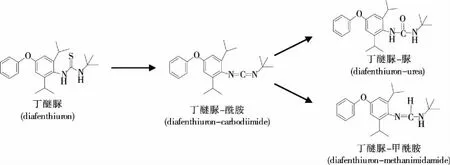
图2 丁醚脲降解规律图Figure 2 The proposed degradation pathways of diafenthiuron
2.4 线性范围、检出限、定量限、回收率和精密度
采用空白基质配制系列不同浓度的混合标准溶液进行测定,以各组分定量离子的峰面积为纵坐标,质量浓度为横坐标绘制工作曲线, 得到的线性回归方程,相关系数r为0.990~0.995(表2),表明各化合物在响应的浓度范围内呈良好的线性关系,外标法定量。以3倍信噪比(S/N)响应时所对应的质量浓度作为检出限(LOD)。以10倍信噪比对应的质量浓度作为定量限(LOQ),经样品添加试验确定丁醚脲、丁醚脲—甲酰胺和丁醚脲—脲的检出限分别为0.005,0.002,0.001 mg/kg,定量限分别为0.010,0.005,0.003 mg/kg。在空白绿茶样品中添加3个不同浓度水平的混合标准溶液进行添加回收试验,每个添加水平平行测定6次,计算回收率和相对标准偏差见表2。结果表明,3个添加水平下各化合物平均回收率为62.2%~99.6%;精密度为1.3%~9.4%(相对标准偏差)。绿茶空白样品及加标样品的多反应监测色谱图见图4。
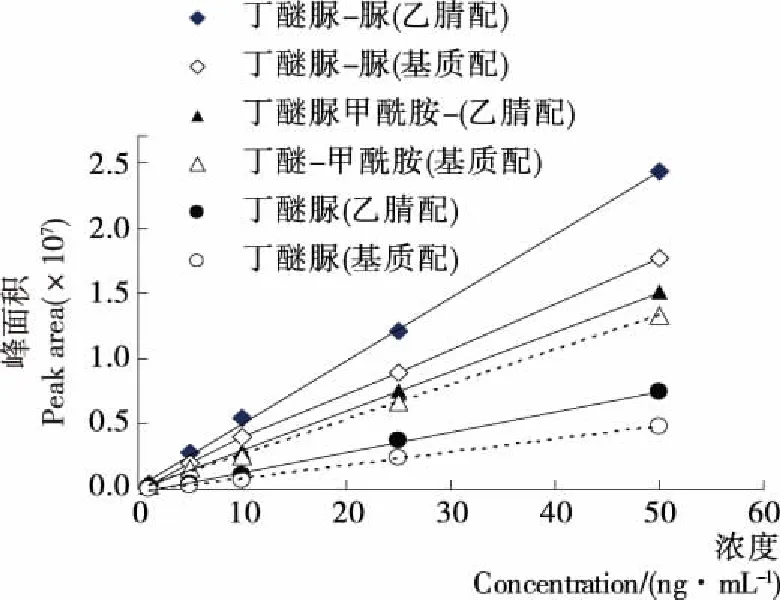
图3 乙腈和空白茶叶样品基质配制混合标准工作 溶液的曲线图
Figure 3 The standard curves of mixed standard prepared with acetonitrile and sample ma trix,respectively
2.5 实际样品测定
应用本方法对457个茶叶样品进行检测,有79个样品同时检出有丁醚脲—甲酰胺和丁醚脲—脲,含量分别为0.008~1.690,0.013~1.320 mg/kg,其中3个样品检出有丁醚脲,含量为0.011~0.054 mg/kg。结果表明丁醚脲在茶叶的残留检测主要以降解产物的形式存在。
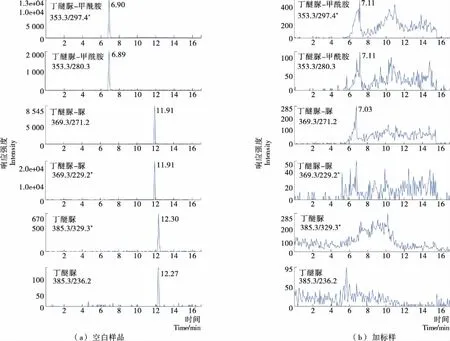
图4 空白样品和添加0.01 mg/kg混合标准品的样品的MRM离子流色谱Figure 4 Multiple reaction monitor(MRM)chromatograms of a blank green tea sample and spiked with standards at 0.01 mg/kg for each compound表2 3种化合物的线性范围、相关系数、定量限、平均回收率及精密度Table 2 Linear range, correlation coefficients (r) , limits of quantitation (LOQ), recoveries and precisions (relative standard deviations,RSDs) of 3 degradation products (n=6)
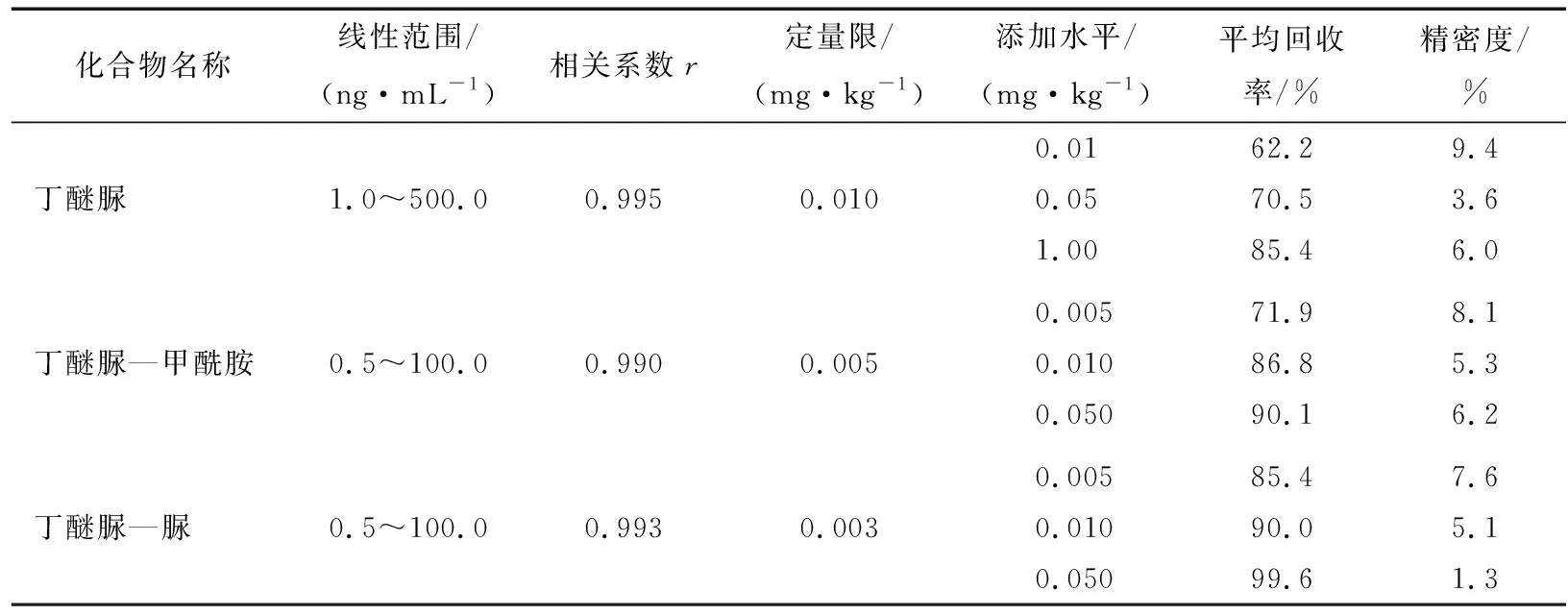
化合物名称线性范围/(ng·mL-1)相关系数r定量限/(mg·kg-1)添加水平/(mg·kg-1)平均回收率/%精密度/%0.0162.29.4丁醚脲1.0~500.00.9950.0100.0570.53.61.0085.46.00.00571.98.1丁醚脲—甲酰胺0.5~100.00.9900.0050.01086.85.30.05090.16.20.00585.47.6丁醚脲—脲0.5~100.00.9930.0030.01090.05.10.05099.61.3
3 结论
利用高效液相色谱—串联质谱法结合QuEChERS前处理技术,建立了茶叶中丁醚脲原药及其2种降解产物残留量的检测方法。该方法定性分析的同时并能对3种化合物进行准确定量,得到丁醚脲及其降解产物的总量,准确反映了茶叶中丁醚脲的实际残留量,满足残留监测的要求。并考察了丁醚脲在溶液中的降解情况,优化前处理方法,有效避免了丁醚脲在前处理过程的降解,回收率稳定且重现性较好。然而丁醚脲在茶叶中的降解机理有待进一步研究。
猜你喜欢
兰州文理学院学报(自然科学版)(2024年2期)2024-06-12 00:00:00
特产研究(2024年1期)2024-03-12 05:40:56
煤化工(2022年3期)2022-07-08 07:24:42
天然产物研究与开发(2019年10期)2019-11-05 10:12:44
现代农业(2016年4期)2016-02-28 18:42:14
中国资源综合利用(2016年10期)2016-01-22 08:36:09
应用化工(2014年1期)2014-08-16 13:34:08
化工生产与技术(2014年3期)2014-02-27 13:41:44
河北医科大学学报(2011年1期)2011-03-25 10:15:30
天津化工(2010年5期)2010-09-18 02:55:58
