
Influence of Electron Donating Ability on Reverse Intersystem Crossing Rate for One Kind of Thermally Activated Delayed Fluorescence Molecules
2018-06-27 06:48:14MinglngWngJinzhongFnLiliLin
Ming-lng Wng,Jin-zhong Fn,Li-li Lin∗
a.Shandong Province Key Laboratory of Medical Physics and Image Processing Technology,School of Physics and Electronics,Shandong Normal University,Jinan 250014,China
b.Department of Electronics,Peking University,Beijing 100871,China
I.INTRODUCTION
Since the milestone work of Tang et al.in 1987,organic light-emitting diodes(OLEDs)have attracted extensive attentions because of their potential application in flat-panel display and solid-state lighting[1−3].In OLEDs,the singlet to triplet exciton formation ratio is 1:3 due to the spin statistics.For normal fluorescence emitters,radiative decay of the triplet excitons that account for 75%is spin forbidden and only the singlet excitons(25%)can be used for light emitting.To realize the goal of fully harvesting the triplet excitons,phosphorescent materials are developed and have achieved great success[4−7].However,the phosphorescent materials are limited to Ir and Pt complexes,thus both fluorescence and phosphorescence OLEDs have advantages and disadvantages.Recently,Adachi et al.successfully achieved 100%internal quantum efficiency(IQE)by the use of pure organic thermally activated delayed fluorescence(TADF)OLEDs[8−12].For effective TADF-OLEDs,a small energy gap(∆ES1-T1)between the lowest singlet excited state(S1)and lowest triplet excited state(T1)is expected,which can be achieved by decreasing the overlap between highest occupied molecular orbital(HOMO)and lowest unoccupied molecular orbital(LUMO).According to the equation,where kBdenotes the Boltzmann constant and T is temperature,a small∆ES1-T1can facilitate the reverse intersystem crossing(RISC)process[13].For improving utilization of excitons,one effective way is to convert triplet excitons into singlet excitons through a rapid RISC process[14−16].Moreover,the spin-orbit coupling coefficient Hsobetween S1and T1is also a key factor that influences the conversion rate,so two important factors Hsoand∆ES1-T1should be determined for realizing high efficient RISC process.
As we know,molecular structures determine their photophysical properties.In order to illustrate the influence of modification in donor groups of TADF molecules on their transition properties,∆ES1-T1,ISC and RISC rates,here we adopt the xanthone(XTN)which is well known for its involved solvent and temperature dependent photophysics as electron acceptor unit[17],the carbazole,phenoxazine(PXZ),9,9-dimethyl-9,10-dihydroacridine(DMAC)and phenothiazine(PTZ)as well as their derivatives which is substituted by diphenylamine as electron donating part to construct carbazole-XTN(a),PXZ-XTN(b),DMACXTN(c),PTZ-XTN(d)as well as carbazole-II-XTN(e),PXZ-II-XTN(f),DMAC-II-XTN(g),PTZ-II-XTN(h),all studied structures are shown in FIG.1.Thus,we can analyze the effect of different electron donating ability and delocalization of frontier molecular orbitals on∆ES1-T1,ISC and RISC rates.Furthermore,we can determine the dominant factor in realizing efficient RISC process and provide some suggestions for designing high efficient TADF emitters.
II.COMPUTATION
The geometry optimizations and frequency calculations are performed for the ground and excited states by using density functional theory(DFT)and timedependent density functional theory(TD-DFT)with the B3LYP functional and 6-31G(d)basis set respectively.No imaginary frequencies are found which can help one to ensure the structure is stabilized.All calculations are carried out by Gaussian 16 package[18].Besides,we not only draw the distribution of HOMO and LUMO but also analyze the overlap between them by Multiwfn(a multifunctional wavefunction analyzer)[19].Moreover,based on the analysis of the excitation component of S1state,the HOMO-LUMO dominates the transition for all studied molecules,so the distribution of HOMO(LUMO)can be represented by the distribution of hole(electron).Moreover,we analyze the delocalization of hole by the following equation Xhole=∫xρhole(r)dr,where x is component of r.The root mean square deviation(RMSD)of hole is used to characterize its distribution breadth.Meanwhile,the coupling coefficients ofandare calculated based on the optimized structures of T1,T2and T3respectively,all results can be acquired by Dalton 2013 package[20].
Finally,the intersystem crossing rate constant from initial singlet/triplet to triplet/singlet states can be calculated based on the perturbation theory as

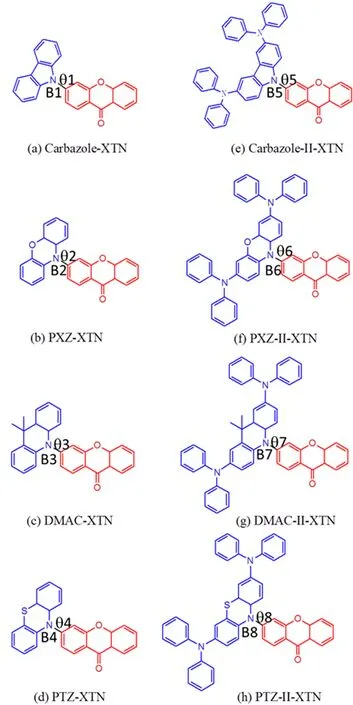
FIG.1 Geometry structures of all studied molecules.
Tif,k(l)is the mixed spin-orbit and non-radiative couplings between two electronic states for the k(l)th normal mode[21,22],

Eq.(6)and Eq.(7)are from Ref.[23]and Ref.[24]respectively.
For the first-order contribution,by applying the thermal vibration correlation function ρIC(t,T),thesimplest and the most commonly employed intersystem crossing rate formalism can be written as:
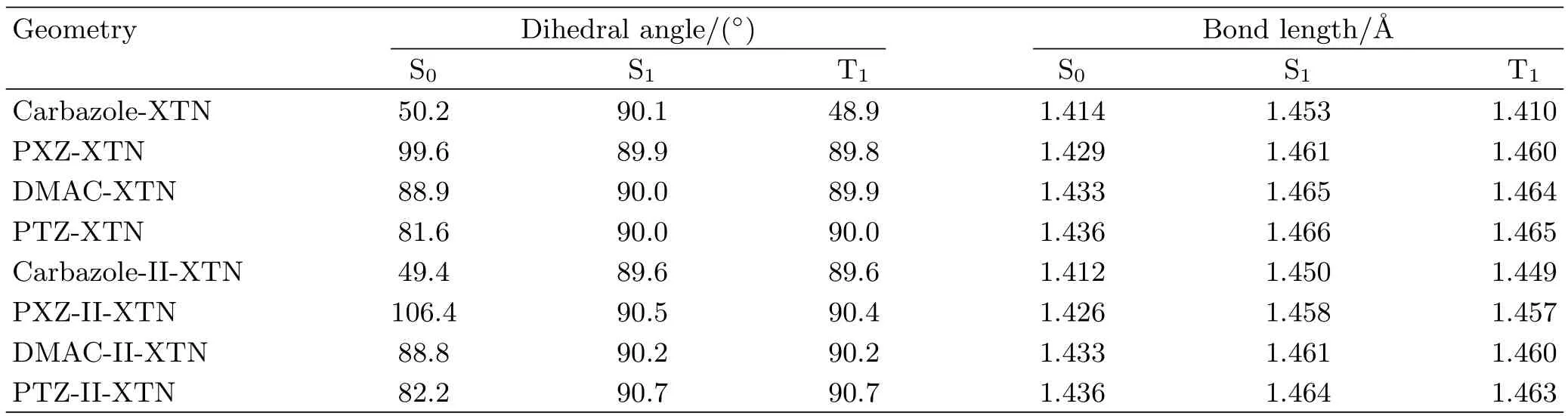
TABLE I Dihedral angle and bond length(marked out in FIG.1)between donor and acceptor for S0,S1and T1are listed respectively based on optimized structures.

All these calculations for ISC and RISC rates are performed by MOMAP(molecular materials property prediction package)promoted by the Institute of Chemistry Chinese Academy of Sciences and Department of Chemistry in Tsinghua University.Both the methodology and application of this formalism can be found in Peng et al’s and Shuai et al’s.works[25−30].
III.RESULTS AND DISCUSSION
A.Geometry structures
According to the method discussed in computational details,the geometry structures of S0,S1and T1for all investigated molecules are optimized by B3LYP functional.Basic molecular structures are shown in FIG.1 and the main geometric parameters are listed in Table I.One can see that different donor units change the dihedral angle and bond length(marked out in FIG.1)between donor and acceptor for S0,S1and T1states.For a more visible comparison,FIG.2 is plotted.Combining Table I and FIG.2,the dihedral angle(θ)is almost unchanged for S1and T1states comparing all studied molecules except for carbazole-XTN,this indicates a small geometry variation when molecule changes from S1state to T1state.Meanwhile,the dihedral angles in S0,S1and T1states are similar for DMAC-XTN and DMAC-II-XTN,this illustrates a small change of reorganization energy from S1to S0and T1to S0.Besides,the bond length between donor and acceptor is slightly changed for S1and T1states compared all studied molecules except for molecule Carbazole-XTN.The bond length of all molecules in S1state is the longest one compared with molecules in S0and T1states,this indicates a decreased interaction between donor and acceptor unit for S1state.Moreover,comparing the bond length of the first four molecules with their diphenylamine substitutions in their S0,S1and T1states respectively,one can see that the latter four molecular bond lengths are reduced comparing with the former four molecules except for the T1states of carbazole-XTN and carbazole-II-XTN.All these results suggest that diphenylamine substitution can decrease the bond length between donor and acceptor unit while little effect on the dihedral angle between them.
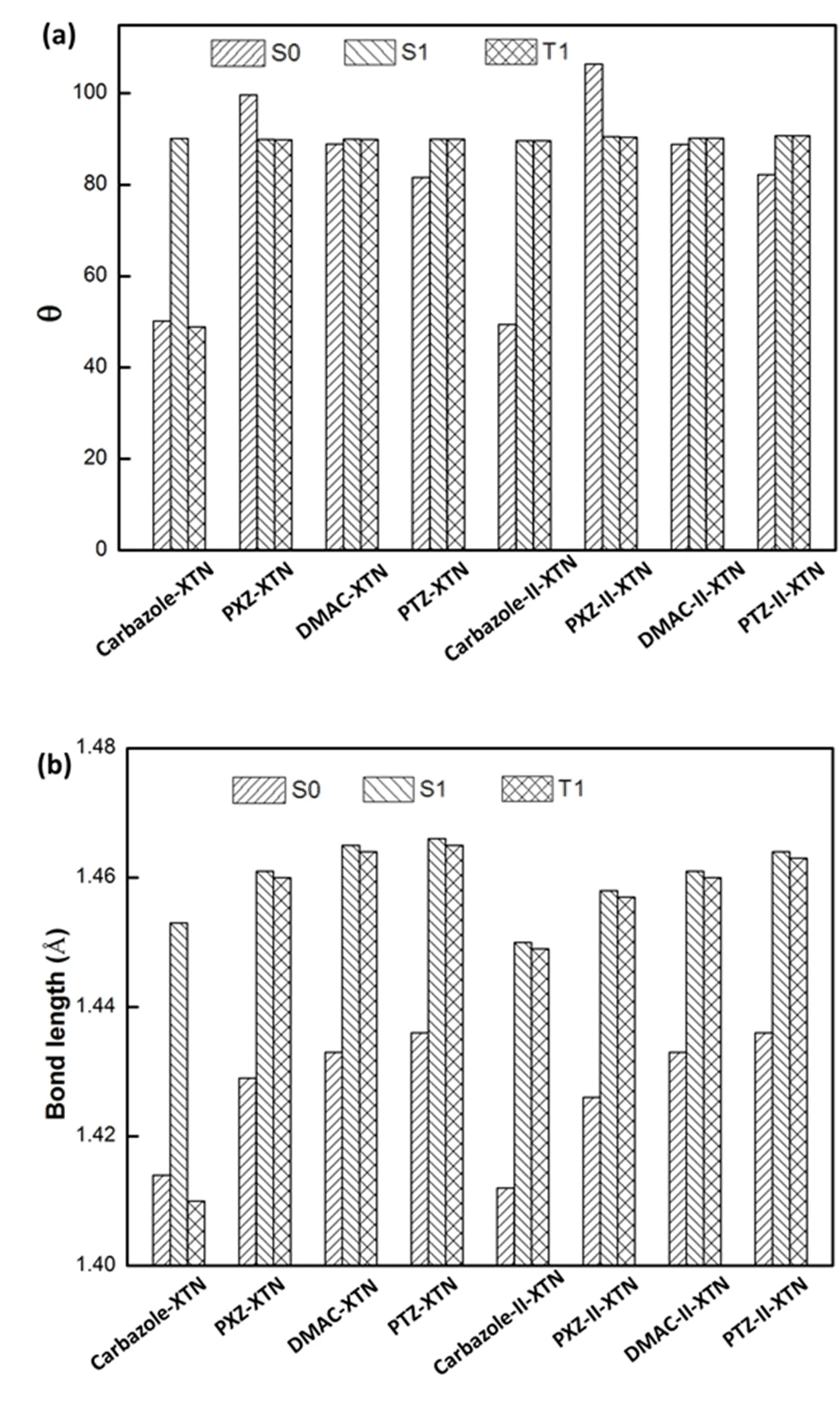
FIG.2 (a)Dihedral angles and(b)bond length between donor and acceptor for optimized S0,S1and T1states of all studied molecules respectively.
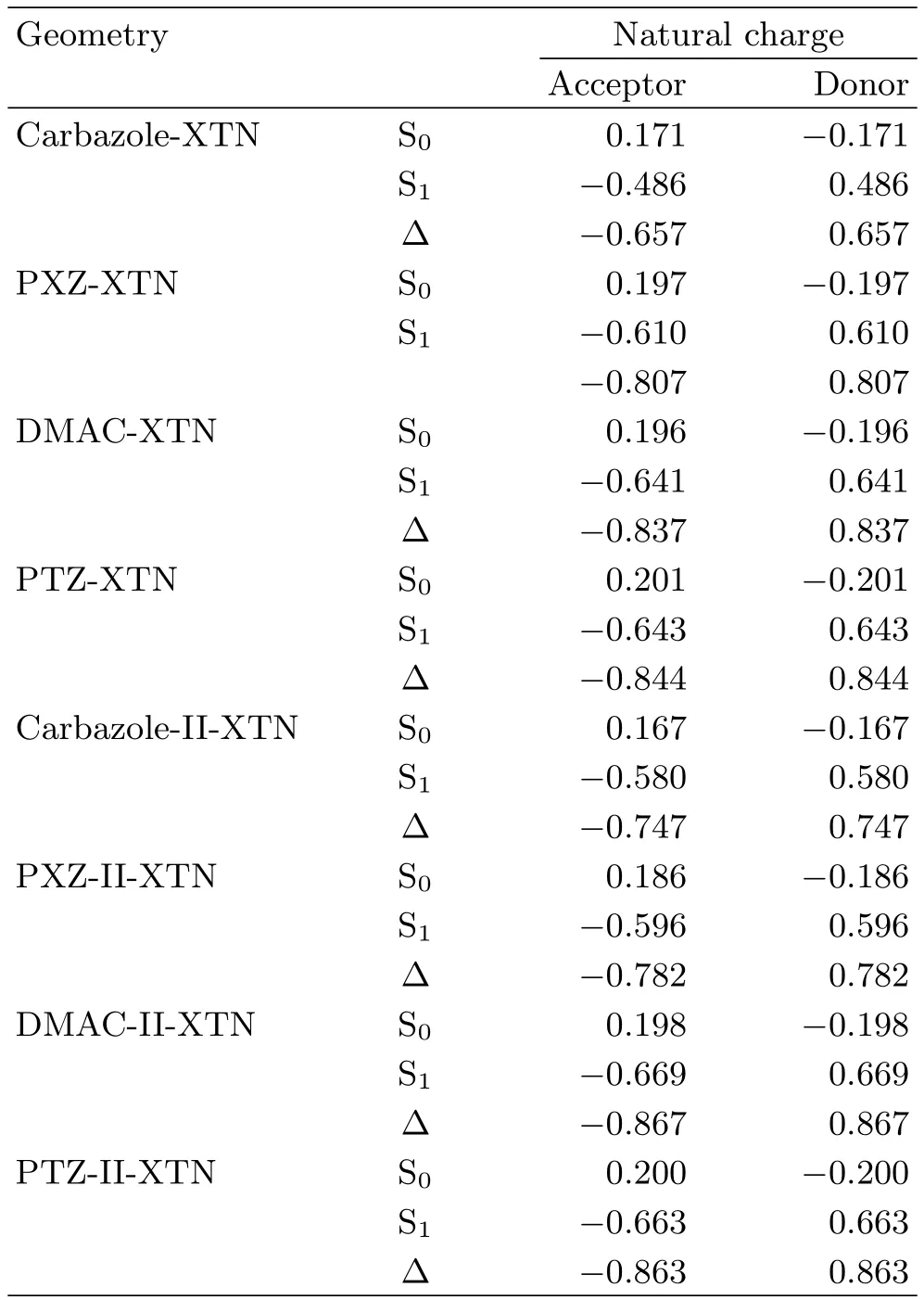
TABLE II Atomic charges of investigated molecules in S0 and S1using NPA method.∆is the charge difference between S1and S0states.
Moreover,the electron-donating ability affects molecular photophysical properties.Atomic charges of the S0and S1states for the eight molecules are calculated by natural population analysis(NPA)method,all data are collected in Table II.From Table II,charges of the donor group for the S0and S1states are negative and positive respectively for all studied molecules,while opposite results are found for the acceptor unit.In addition,the charge difference(∆)between S1and S0is calculated,we use the value of∆to measure the electron donating ability,the larger the charge difference is,the stronger the electron-donating ability is.Thus,we illustrate the effect of different electron-donating to∆ES1-T1and transition properties.Moreover,comparing the former four molecules and the corresponding ones with diphenylamine added in donor unit,the effect of delocalization of molecular orbital to photophysical properties can be analyzed.
B.Frontier molecular orbital properties
Composition of frontier molecular orbital(FMO)is closely related to the molecular excitation propertiessuch as absorption and emission properties.Moreover,ultrafast excited state dynamics investigation is a research hotspot[31,32].In order to get a deep understanding of photophysical behavior of all investigated compounds,analysis of FMO at S0state is performed.Distributions of HOMO and LUMO as well as their energy levels are plotted in FIG.3.One can see that the HOMO and LUMO are localized in donor and acceptor unit respectively,and small orbital overlap between HOMO and LUMO is found.According to the following equation

TABLE III Overlap between HOMO and LUMO(S)as well as the value of RMSD of hole(δhole)and electron(δelectron)with the unit of˚A are listed.

a small∆ES1-T1can be expected.Moreover,the delocalization of frontier orbitals should also be considered.It is reasonable to obtain the same overlap between the HOMO and LUMO for two different molecules such as,one which has the electronic density of both orbitals confined to one group of the molecule and a second for which the density of both orbitals is delocalized over the whole molecular sca ff old.This degree of spatial confinement is important.Comparing molecule Carbazole-XTN(a)with carbazole-II-XTN(e),PXZ-XTN(b)with PXZ-II-XTN(f),DMAC-XTN(c)with DMAC-IIXTN(g)and PTZ-XTN(d)with PTZ-II-XTN(h),one can see that the energy of LUMO is almost unchanged while the energy of HOMO is increased,which brings a decreased HOMO-LUMO energy gap for later four molecules.Moreover,the diphenylamine in donor part not only adjusts the HOMO-LUMO energy level but also increases the delocalization of HOMO.In order to achieve quantitative comparison,the index of S,δholeand δelectronare used to characterize the HOMO-LUMO overlap as well as the delocalization of HOMO and LUMO respectively,all calculated data are collected in Table III.Furthermore,we analyze the electrondonating ability(∆)due to its role in determining the molecular orbital properties.Relationship between S and∆is shown in FIG.4.An inversely proportional relationship is graphed,namely,the stronger the electron-donating ability is,the smaller the HOMO-LUMO overlap is.Through comparing the value of δholefor later four molecules with the former four molecules,the value of δholeis increased when diphenylamine is added in donor unit,so the later four molecules possess larger delocalization of HOMO.For the former four molecules,the δelectrondecreases((a)>(b)>(c)>(d))with the donating ability increases((a)<(b)<(c)<(d)).While for the later four molecules,similar condition is found with the donating ability is(g)≈(h)>(f)>(e)and the δelectronis(h)≈(g)<(f)<(e).Thus,an effective way to decrease∆ES1-T1is illustrated that either to increase the electron donating ability or enlarge the delocalization of HOMO can bring a small∆ES1-T1.

FIG.3 Calculated energy levels,energy gaps(in eV),and orbital composition distributions of the HOMO and LUMO for all molecules(isovalue=0.02).
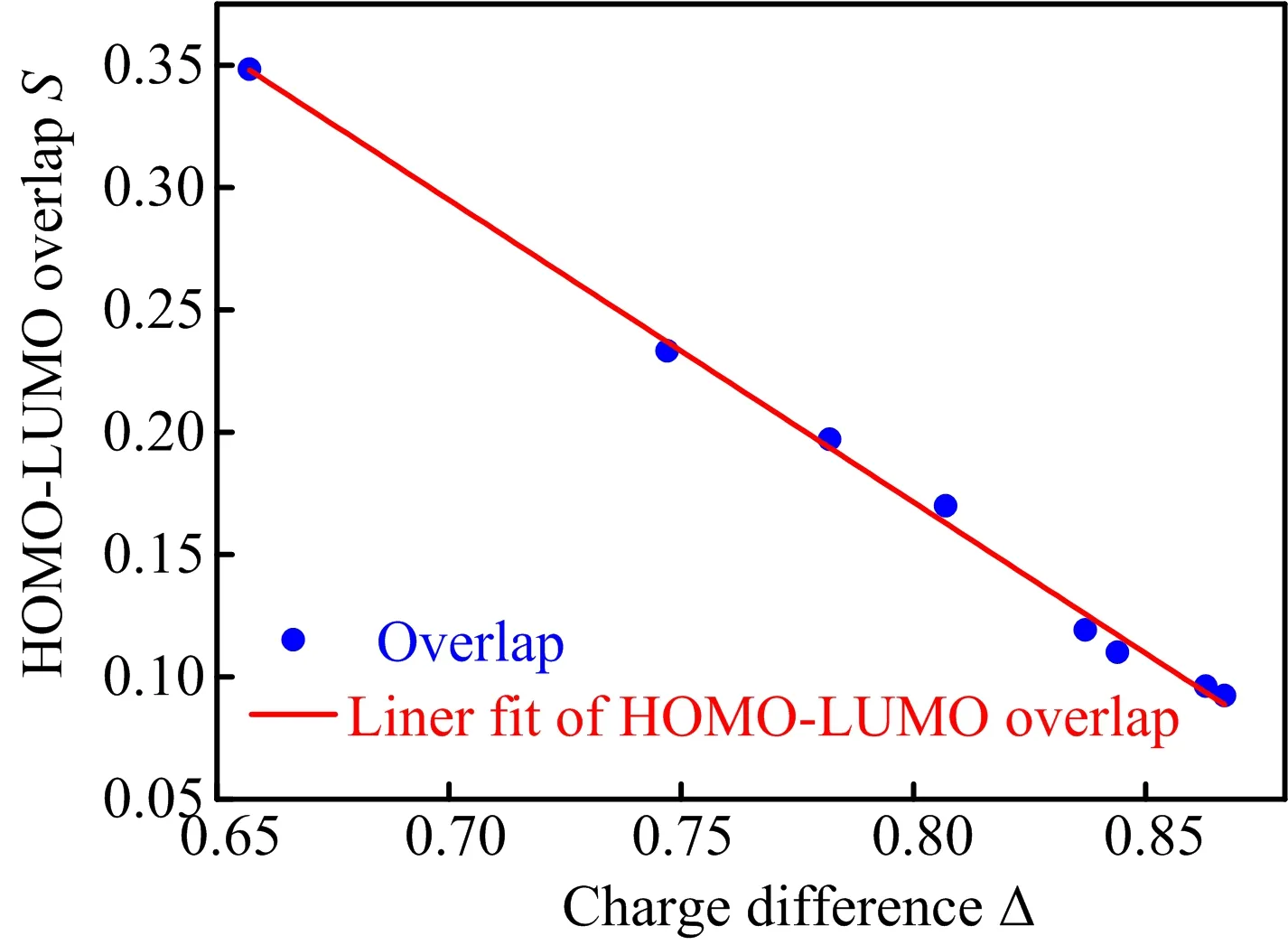
FIG.4 Relationship between charge difference(∆)and HOMO-LUMO overlap(S).
In order to determine the dominant factor in decreasing the∆ES1-T1,relationship between HOMO-LUMO overlap,delocalization of molecular orbital and∆ES1-T1is analyzed.Values of∆ES1-T1for all studies molecules are calculated by TD-DFT method through optimizing excited state geometries,and the adiabatic excitation energies of S1and T1are corrected by zero point vibrational energy(ZPVE).All data are collected in Table IV,and we elaborate the effect of HOMO-LUMO overlap and delocalization of HOMO on∆ES1-T1.Comparing the value of S,δholeand∆ES1-T1between carbazole-XTN(a)and carbazole-II-XTN(e),DMAC-XTN(c)and DMAC-II-XTN(g)as well as PTZ-XTN(d)and PTZ-II-XTN(h),one know that the S is decreased,while the δholeis increased for molecules with diphenylamine added in donor unit,and a decreased∆ES1-T1is obtained.While for PXZ-XTN(b)and PXZ-IIXTN(f),S and δholeare all increased,and a decreased∆ES1-T1is also found.This means that the additional diphenylamine in donor part can decrease∆ES1-T1.Through above-mentioned comparisons,we can come to the conclusion that the enlarge delocalization of molecular orbitals with large separation between HOMO and LUMO can bring a small∆ES1-T1.
C.Transition properties
In order to investigate the electronic transition nature of all studied compounds,TD-DFT calculations are performed based on their optimized S0states.The vertical excitation energy of S1(EVA(S1)),T1(EVA(T1))and their gaps(Evert)as well as the adiabatic excitation energies of S1(E0-0(S1)),T1(E0-0(T1))and theirgaps(∆ES1-T1)are all collected in Table IV.Results show that the value of Evertis inversely proportional to electron-donating ability,the stronger the electrondonating ability is,the smaller the Evertis.Moreover,we calculate the energy landscape of single and triplet states to determine the intersystem crossing and reverse intersystem crossing processes,and their transition properties are analyzed by natural transition orbital(NTO)method.As shown in FIG.5,the energy of T1is lower than S1and no extra energy levels are found between them for studied molecules except for carbazole-XTN.S1and T1states all possess charge transfer(CT)properties which can facilitate the reverse conversion from T1to S1[33,34].For carbazole-XTN,T2and T3are also lower than S1,thus the ISC and RISC processes occur between S1and T2as well as S1and T3.Moreover,T2and T3possess localized excitation(LE)properties,this feature can also affect the ISC and RISC processes[35].Corresponding data are shown in the following section.
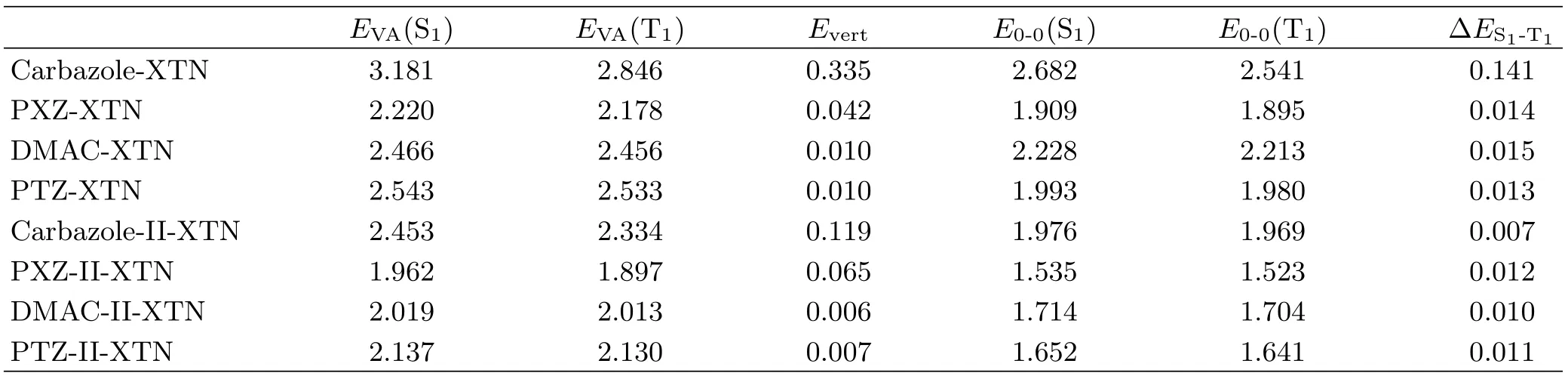
TABLE IV Vertical excitation energies of S1(EVA(S1)),T1(EVA(T1))and their gaps(Evert)as well as their adiabatic excitation energies(with ZPVE correction)of S1(E0-0(S1))and T1(E0-0(T1))and their gaps(∆ES1-T1).Units are in eV.

TABLE V Spin-orbit coupling constants(cm−1)between S1and T1for studied molecules.
D.ISC and RISC rates
As we all know,ISC and RISC processes play a crucial role in efficient TADF-OLEDs.Through abovementioned results,we know that∆ES1-T1is largely dependent on the frontier orbital overlap and delocalization.The transition nature of S1and T1(T2or T3)also plays an important role in achieving efficient ISC and RISC processes.In order to calculate the ISC and RISC rate parameters,spin-orbit coupling coefficients(Hso)between singlet and triplet states are acquired by Dalton 2013 package,corresponding data are collected in Table V.Further,the ISC and RISC rates between S1and T1are calculated by MOMAP package,and results are summarized in Table VI.From Table V and Table VI,one can see that Hsoof carbazole-XTN between S1and T1is the biggest one(1.75 cm−1)among all studied molecules,but the ISC and RISC rates are smaller than that of remainders,this is due to its large∆ES1-T1(0.141 eV).So we investigate the ISC and RISC processes between S1and T2as well as S1and T3for carbazole-XTN,and all data are summarized in Table VII.For carbazole-XTN,the spinorbit coupling coefficientsare larger than that ofwith decreased adiabatic energy gap.However,the ISC and RISC rates between S1and T3are comparable with these between S1and T1,this is related to the LE transition nature of T3.Gibson and Penfold found that3LE often brings a stable triplet state while3CT can promote the reverse conversion from3CT to1CT[36].Furthermore,we investigate the relationship between spin-orbit coupling coefficient Hso,∆ES1-T1,and RISC rate for studied molecules except for the carbazole-XTN,corresponding results are shown in FIG.6.A liner relationship
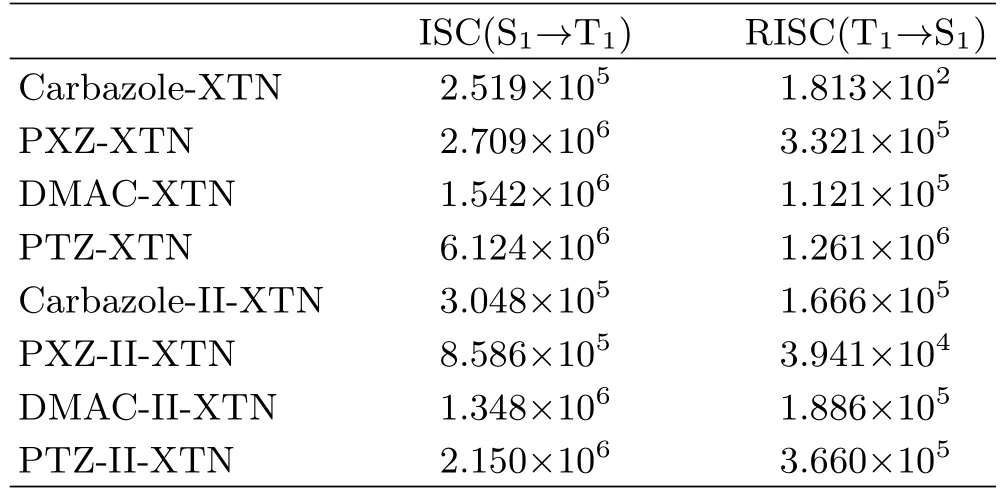
TABLE VI The calculated intersystem crossing rate and reverse intersystem crossing rates for all molecules with the unit of s−1.

FIG.5 The energy levels and molecular orbital characters of different excited states for all studied molecules.The value above every arrow represents the ratio in the corresponding transition.
TABLE VII Spin-orbit coupling constants⟩andas well as the ISC and RISC rates are listed with the unit of s−1.

TABLE VII Spin-orbit coupling constants⟩andas well as the ISC and RISC rates are listed with the unit of s−1.
⟨S1|ˆHso|T2⟩5.888 ISC(S1→T2)1.339×106⟨S1|ˆHso|T3⟩2.307 ISC(S1→T3)8.274×103 RISC(T2→S1)3.434×105 RISC(T3→S1)7.296×102
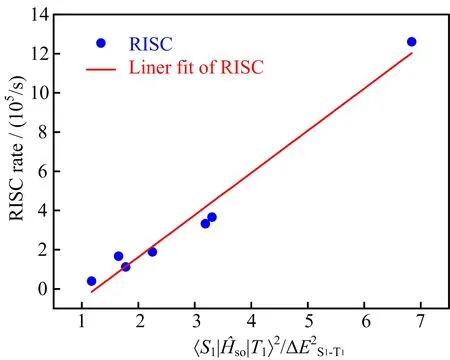
FIG.6 Relationship betweenand RISC rate.

IV.CONCLUSION
In this work,the electronic structures,molecular orbital properties,energy gaps,excitation properties and RISC process of all eight molecules are investigated by DFT and TDDFT methods.Through our investigations,the diphenylamine substitution in the donor unit has little effect on the dihedral angle between donor and acceptor unit,but can decrease the bond length between them except for the T1state of Carbazole-XTN.The electron donating ability,HOMO-LUMO overlap and frontier molecular orbital delocalization are quantitatively calculated.Results show that the overlap between HOMO and LUMO is decreased when the electron donating ability of donor groups is increased.As the diphenylamine groups are added in donor part,the delocalization of HOMO is enlarged,this brings a decreased energy gap between S1and T1state.Moreover,the spin-orbit coupling coefficient plays a significant role in realizing high efficient RISC process,large value ofcan accelerate the exciton conversion from T1to S1.All our investigated molecules possess small∆ES1-T1and fast RISC rates,these molecules can be regarded as promising candidates for efficient TADF molecules.Furthermore,a wise molecular design strategy that enlarges the delocalization of frontier molecular orbitals with large separation between HOMO and LUMO,is proposed to achieve a small∆ES1-T1.
V.ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China(No.11374195 and No.21403133),the Taishan Scholar Project of Shandong Province,the Promotive Research Fund for Excellent Young and Middle-aged Scientists of Shandong Province(No.BS2014CL001),and the General Financial Grant from the China Postdoctoral Science Foundation(No.2014M560571).Great thanks to Professor Yi Luo at University of Science and Technology of China,Professor Zhi-gang Shuai at Tsinghua University and Qian Peng at Institute of Chemistry,Chinese Academy of Sciences for their helpful suggestions in our calculation.Thanks to Professor Ying-li Niu at Beijing Jiaotong University for his great help in the usage of MOMAP.
[1]C.W.Tang and S.A.VanSlyke,Appl.Phys.Lett.51,913(1987).
[2]J.H.Jou,S.Kumar,A.Agrawal,T.H.Li,and S.Sahoo,J.Mater.Chem.C 3,2974(2015).
[3]S.Xu,R.F.Chen,C.Zheng,and W.Huang,Adv.Mater.28,9920(2016).
[4]C.Adachi,M.A.Baldo,M.E.Thompson,and S.R.Forrest,J.Appl.Phys.90,5048(2001).
[5]Z.Kuang,X.Wang,Z.Wang,G.He,Q.Guo,L.He,and A.Xia,Chin.J.Chem.Phys.30,259(2017).
[6]C.Li,L.Duan,D.Zhang,and Y.Qiu,Acs.Appl.Mater.Inter.7,15154(2015).
[7]S.Cao,L.Hao,W.Y.Lai,H.Zhang,Z.Yu,X.Zhang,X.Liu,and W.Huang,J.Mater.Chem.C 4,4709(2016).
[8]H.Uoyama,K.Goushi,K.Shizu,H.Nomura,and C.Adachi,Nature 492,234(2012).
[9]J.Guo,X.L.Li,H.Nie,W.Luo,R.Hu,A.Qin,Z.Zhao,S.J.Su,and B.Z.Tang,Chem.Mater.29,3623(2017).
[10]J.Guo,X.L.Li,H.Nie,W.Luo,S.Gan,S.Hu,R.Hu,A.Qin,Z.Zhao,S.J.Su,and B.Z.Tang,Adv.Funct.Mater.27,1606458(2017).
[11]L.Yu,Z.Wu,C.Zhong,G.Xie,K.Wu,D.Ma,and C.Yang,Dyes.Pigments.141,325(2017).
[12]J.Luo,S.Gong,T.Zhang,C.Zhong,G.Xie,Z.H.Lu,and C.Yang,Dyes.Pigments.147,350(2017).
[13]T.Sato,M.Uejima,K.Tanaka,H.Kaji,and C.Adachi,J.Mater.Chem.C 3,870(2015).
[14]Q.S.Zhang,H.Kuwabara,W.J.Potscavage Jr.,S.P.Huang,Y.Hatae,T.Shibata,and C.Adachi,J.Am.Chem.Soc.136,18070(2014).
[15]J.Z.Fan,S.Qiu,L.L.Lin,and C.K.Wang,Chin.J.Chem.Phys.29,291(2016).
[16]J.Fan,L.Cai,L.Lin,and C.Wang,Chem.Phys.Lett.664,33(2016).
[17]M.Y.Wong and E.Zysman-Colman,J.Mater.Chem.C 29,1605444(2017).
[18]M.J.Frisch,G.W.Trucks,H.B.Schlegel,G.E.Scuseria,M.A.Robb,J.R.Cheeseman,G.Scalmani,V.Barone,B.Mennucci,G.A.Petersson,H.Nakatsuji,M.Caricato,X.Li,H.P.Hratchian,A.F.Izmaylov,J.Bloino,G.Zheng,J.L.Sonnenberg,M.Hada,M.Ehara,K.Toyota,R.Fukuda,J.Hasegawa,M.Ishida,T.Nakajima,Y.Honda,O.Kitao,H.Nakai,T.Vreven,J.A.Montgomery Jr.,J.E.Peralta,F.Ogliaro,M.Bearpark,J.J.Heyd,E.Brothers,K.N.Kudin,V.N.Staroverov,R.Kobayashi,J.Normand,K.Raghavachari,A.Rendell,J.C.Burant,S.S.Iyengar,J.Tomasi,M.Cossi,N.Rega,N.J.Millam,M.Klene,J.E.Knox,J.B.Cross,V.Bakken,C.Adamo,J.Jaramillo,R.Gomperts,R.E.Stratmann,O.Yazyev,A.J.Austin,R.Cammi,C.Pomelli,J.W.Ochter-ski,R.L.Martin,K.Morokuma,V.G.Zakrzewski,G.A.Voth,P.Salvador,J.J.Dannenberg,S.Dapprich,A.D.Daniels,Ö.Farkas,J.B.Foresman,J.V.Ortiz,J.Cioslowski,and D.J.Fox,Gaussian 16,Revision A.03,Wallingford CT,USA:Gaussian Inc.(2016).
[19]T.Lu and F.W.Chen,J.Comput.Chem.33,580(2012).
[20]Dalton,a Molecular Electronic Structure Program,http://daltonprogram.org.
[21]Y.L.Niu,Q.Peng,C.M.Deng,X.Gao,and Z.G.Shuai,J.Phys.Chem.A 114,7817(2010).
[22]Q.Peng,Q.H.Shi,Y.L.Niu,Y.P.Yi,S.R.Sun,W.Li,W.Q,and Z.G Shuai,J.Mater.Chem.C.4,6829(2016).
[23]Q.Peng,Y.L.Niu,Q.H.Shi,X.Gao,and Z.G.Shuai,J.Chem.Theory.Comput.9,1132(2013).
[24]T.Zhang,H.L.Ma,Y.L.Niu,W.Q.Li,D.Wang,Q.Peng,Z.G.Shuai,and W.Z.Liang,J.Phys.Chem.C 119,5040(2015).
[25]Z.G.Shuai and Q.Peng,Phys.Rep.537,123(2014).
[26]J.Z.Fan,L.Cai,L.L.Lin,and C.K.Wang,J.Phys.Chem.A 120,9422(2016).
[27]J.Fan,L.Lin,and C.K.Wang,J.Mater.Chem.C 5,8390(2017).
[28]L.Lin,Z.Wang,J.Fan,and C.Wang,Org.Electron.41,7(2017).
[29]J.Fan,L.Lin,and C.K.Wang,Phys.Chem.Chem.Phys.19,30147(2017).
[30]J.Fan,L.Cai,L.Lin,and C.K.Wang,Phys.Chem.Chem.Phys.19,29872(2017).
[31]X.C.Li,N.Sui,Q.H.Liu,Q.L.Yuan,and Y.H.Wang,Chin.J.Chem.Phys.29,389(2016).
[32]Y.P.Wang,S.Zhang,S.m.Sun,K.Liu,and B.Zhang,Chin.J.Chem.Phys.26,651(2013).
[33]M.K.Etherington,J.Gibson,H.F.Higginbotham,T.J.Penfold,and A.P.Monkman,Nat.Commun.7,13680(2016).
[34]Y.Gao,S.Zhang,Y.Pan,L.Yao,H.Liu,Y.Guo,Q.Gu,B.Yang,and Y.Ma,Phys.Chem.Chem.Phys.18,24176(2016).
[35]J.Gibson,A.P.Monkman,and T.J.Penfold,ChemPhysChem 17,2956(2016).
[36]J.Gibson and T.J.Penfold,Phys.Chem.Chem.Phys.19,8428(2017).
 CHINESE JOURNAL OF CHEMICAL PHYSICS2018年3期
CHINESE JOURNAL OF CHEMICAL PHYSICS2018年3期
- CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Network Modeling of Inflammatory Dynamics Induced by Biomass Smoke Leading to Chronic Obstructive Pulmonary Disease
- A Double Network Hydrogel with High Mechanical Strength and Shape Memory Properties
- A High-Performance and Flexible Chemical Structure&Data Search Engine Built on CouchDB&ElasticSearch
- Nucleation of Boron-Nitrogen on Transition Metal Surface:A First-Principles Investigation
- Maximum Thermodynamic Electrical Efficiency of Fuel Cell System and Results for Hydrogen,Methane,and Propane Fuels
- Electronic Structure and Optical Properties of K2Ti6O13Doped with Transition Metal Fe or Ag