
Role of Nanocavity Plasmons in Tunneling Electron Induced Light Emission on and near a Molecule
2018-06-27 06:48:06HuifngWngGongChenXiogungLiZhenhoDong
Hui-fng Wng,Gong Chen,b∗,Xio-gung Li,Zhen-ho Dong∗
a.Hefei National Laboratory for Physical Sciences at the Microscale,University of Science and Technology of China,Hefei 230026,China
b.School of Physics and Engineering,Zhengzhou University,Zhengzhou 450052,China
c.Institute for Advanced Study,Shenzhen University,Shenzhen 518060,China
I.INTRODUCTION
Scanning tunneling microscope(STM)offers a unique platform for characterizing and manipulating lightmatter interactions at the atomic scale.By controlling charge injection with sub-nanometer spatial precision and simultaneously recording the frequency-resolved emission spectrum,the electronic and vibrational properties of a single molecule can be revealed[1–3].It is well recognized that nanocavity plasmons(NCP)play an important role in STM induced molecular emission,including enhancing the emission intensity,reshaping the spectral profile and even generating new emission channels[3–10].
Nevertheless,the role of NCP in the excitation process of molecules is still under debate.For example,while it is proposed that light emission from molecules can be attributed to resonant carrier injections into molecular orbitals[1,11]and the major role of plasmons is to enhance the emission rate rather than to directly excite the emitter[6–8],it is also argued that the NCP excited by inelastic tunneling electrons can act as a local light source to excite the molecules via their mutual electromagnetic interactions[5,12].However,
in recent experiments where the STM tip was placed in close proximity to the edge of a single molecule to prevent direct carrier injection into the molecule and therefore only the NCP can be excited by the tunneling electrons initially,only molecule-modified NCP emission with asymmetric Fano dips,rather than sharp molecular peaks,were observed[13,14].Consequently,the role of NCP in STM induced light emission from molecular junctions still remains to be further studied.In this paper,we study theoretically the time evolution and spectral properties of the emission from the coupled plasmon-molecule system using time-dependent quantum master equations.To mimic STM induced luminescence(STML)experiments,a single molecule and a single-mode NCP are assumed to be selectively excited initially,depending on the tip position with respect to the molecule.The effect of NCP on the molecular emission and the effect of individual molecules on the NCP emission will be studied in detail.The influence of the magnitude of the radiative decay rate of molecules on the emission spectral profile will also be discussed briefly.Our model helps to clarify the role of plasmon-molecule interactions in affecting the spectroscopic properties of biased molecular junctions.
II.THEORETICAL MODEL
The master equation describing the dynamics of the coupled plasmon-molecule system can be written as[5,7,8]
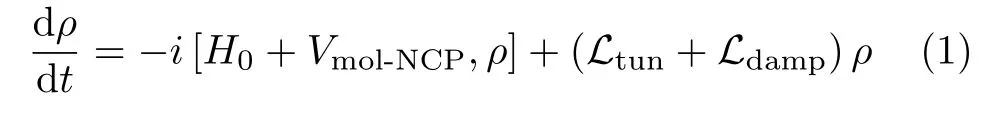
where H0=Hmol+HNCPis the Hamiltonian for the coupling-free molecule and NCP,ρ=ρmol⊗ρNCPis the density operator for the coupled plasmon-molecule system.The interactions between the single molecule and the NCP are considered in the dipole approximationwhereis the creation(destruction)operator for the NCP,is the excitation(de-excitation)operator for the molecular exciton,andare the ground and excited states of the molecule,respectively.For simplicity,only a single-mode NCP is taken into account.Ltunand Ldampare Liouvillian operators that account for tunneling currents induced excitation and various damping processes,respectively,which will be detailed below.
The Liouvillian operator Ltunaccounts for the tunneling events with either the molecule or the NCP excited[5,7,8]
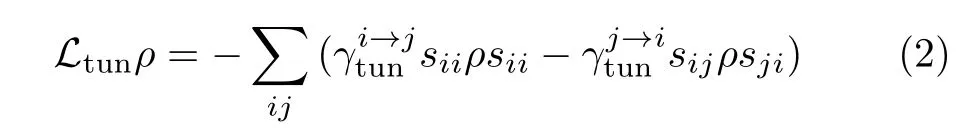
whereis the tunneling electron induced transition rate from the i-th level to the j-th level of the plasmonmolecule system,sijis a square matrix with the element(i,j)equaling to 1 while all other elements vanish.
The Liouvillian operator Ldampaccounts for various damping processes and takes the standard Lindblad form[5,7,8]
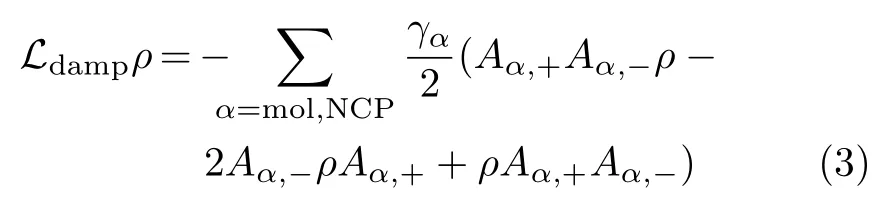
whereand I is a unit operator.γmoland γNCPrepresent the total(radiative plus non-radiative)decay rate of the molecule and the NCP,respectively.
After solving the time-dependent master equation,the density operator ρ(t)that describes the emission properties of the coupled system can be obtained[15].The populations of the molecular exciton Pmol(t)and the NCP PNCP(t),corresponding to the diagonal elements of the density matrix,are evaluated as
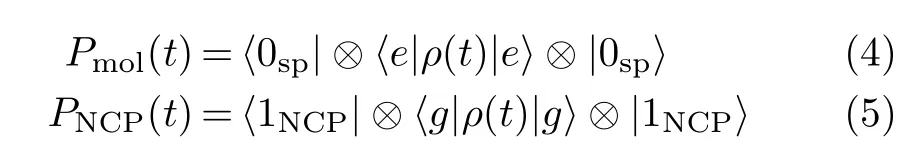
Furthermore,to better understand the light emission properties of the coupled plasmon-molecule system,the total emission is artificially decomposed into two parts[16],i.e.,the light emission from the single molecule
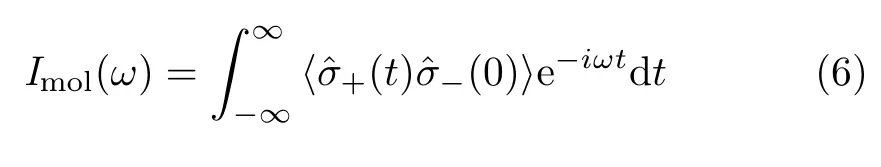
and the light emission from the NCP

These two time correlation functions of the molecular and plasmonic operators are calculated via the quantum regression theorem[17].
III.RESULTS AND DISCUSSION
To make the numerical results more realistic,the experimental parameters in Ref.[13]were adopted in the present work.Specifically,the total decay rates of the electronically decoupled molecule and the NCP in STM junctions are estimated to be γmol≈15 meV and γNCP≈160 meV,respectively.The plasmon-molecule coupling strength g depends on the lateral distance between the tip apex and the molecule,and a typical value when the tip apex is close to the edge of the molecule reported in Ref.[13]is g≈15 meV.The transition dipole moment of a single molecule used in STML is typically a few Debye,corresponding to a radiative decay rate ofeV in vacuum[7].According to the above parameters,the spontaneous emission rate of a molecule in the STM junction has been increased to∼4g2/γNCP≈5.6×10−3eV.Compared with the radiative decay rate of the same molecule in vacuum(8×10−8eV)[13],such a large enhancement corresponds to a large Purcell factor of∼105,which clearly demonstrates that the spontaneous emission rate of the molecule is enormously enhanced due to its coupling to the NCP.However,it is difficult to obtain the radiative decay rate of the NCP directly from experiments.Nevertheless,Persson and Barato ff[18]have shown theoretically that the quantum efficiency for the emission of localized surface plasmons is∼0.1 for a spherical nanoparticle with a radius of a few tens of nanometers,sois set to be 16 meV.
In the following,we shall discuss time-dependent populations and emission intensities as well as emission spectral properties of the coupled plasmon-molecule system under selective initial excitation of either the molecule or the NCP.In STM induced light emission experiments from a single molecule,the tunneling currents used are typically sub-nanoamperes,corresponding to an average time interval between two successive tunneling electrons on a time scale of nanoseconds[1–3],which is much longer than the lifetime of either the NCP or the molecular exciton near a plasmonic nanocavity on a time scale of sub-picoseconds.Therefore,when evaluating the time-dependent populations of the coupled system,we can start from a specified initial state at t=0 and ignore the tunneling electron induced excitation term(namely,the Ltunterm in Eq.(1)).Specifically,FIG.1(b)and(c)are obtained with the initialwith only the molecule excited,and FIG.2(b)and(c)are obtained with the initial state|g⟩⊗|1NCP⟩with only the NCP excited.On the other hand,when evaluating the steady-state emission spectrum in FIG.1(d)and FIG.2(d),the tunneling electron induced excitation term in Eq.(1)has also been taken into account,specifically calculated by using Eq.(2).
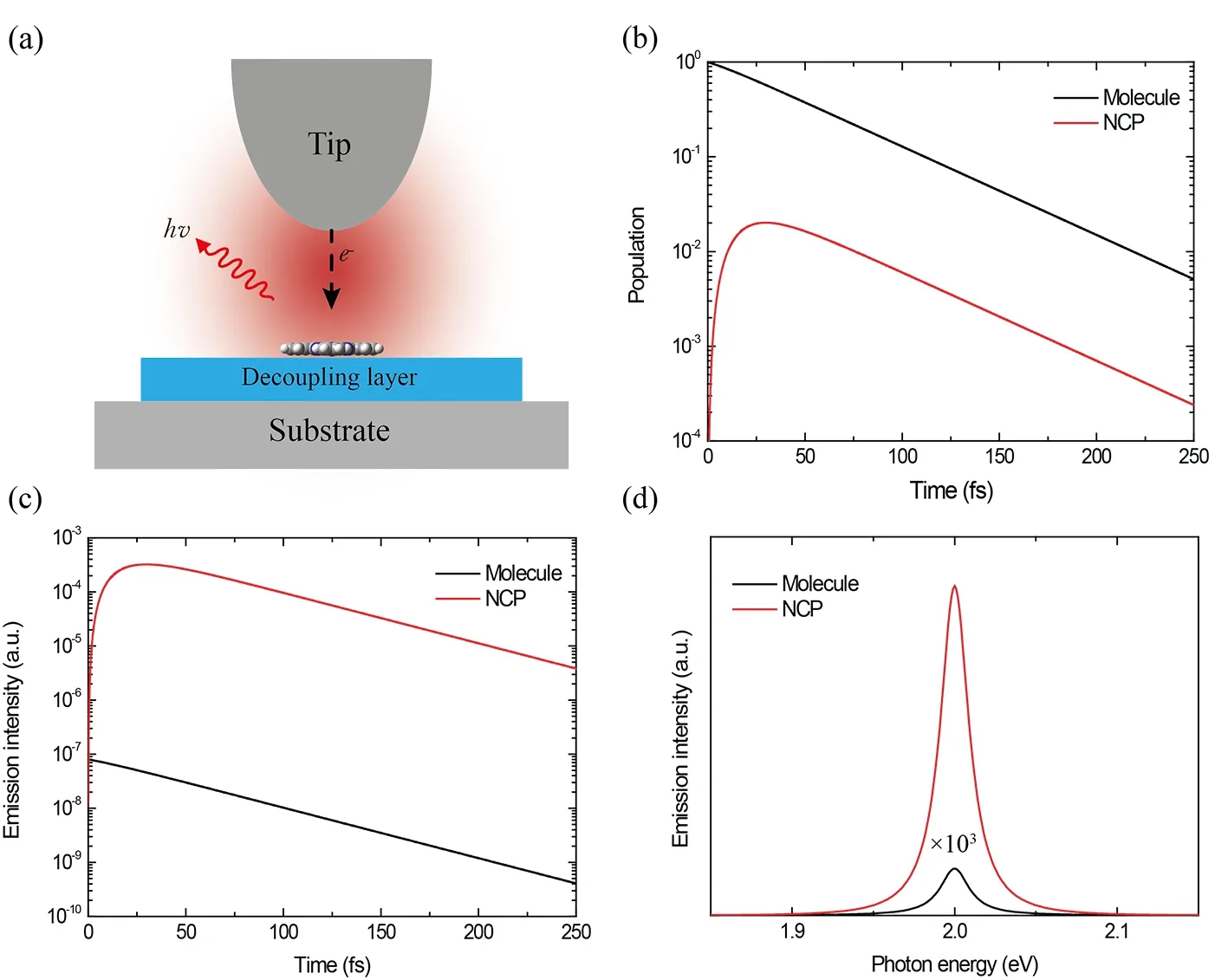
FIG.1 (a)Schematic of STM induced molecular emission with the tip apex positioned above a molecule that can be directly excited by tunneling electrons.(b)and(c)show the time evolution of the populations(b)and emission intensities(c)of the NCP and the molecule,respectively,after the molecule is initially excited.(d)Emission spectra for the molecule and the NCP when the molecule is selectively excited.The emission energy for both the molecule and the NCP is set to 2 eV.
As shown in FIG.1(a),when the STM tip is placed on an electronically decoupled molecular emitter,the tunneling electrons may trigger an exciton in the molecule,whose time evolution is subjected to both the plasmonmolecule coupling and the various radiative and nonradiative decay channels.Without the NCP,the radiative emission power will be simply determined by the state population of the molecular exciton as Wmol(t)=Pmol(t)Due to the presence of the NCP,an additional emission channel is established with the energy transferred to NCP and then emitted to far field.That is,although initially only the molecule is excited,the excitation energy will be transferred between the molecule and the NCP due to their mutual interactions.The time-dependent populations of the molecular exciton(Pmol(t))and the NCP(PNCP(t))for a given coupling strength are plotted in FIG.1(b),which shows that the excitation energy is rapidly transferred from the molecular exciton to the NCP whose population quickly increases from PNCP=0 at t=0 to a maximum of PNCP=0.02 at t≈30 fs.After reaching its maximum,the NCP population then decreases monotonically with time;this is because as time increases,the excitation energy in the molecule is gradually lost and therefore it cannot compensate for the ultrafast NCP dissipation.To quantitatively compare the emission directly from the molecule with that mediated by the NCP,their instantaneous emitting power,determined by the product of the population and the corresponding radia-are plotted in FIG.1(c).Since the radiative decay rate of the NCP is orders of magnitude larger than that of a single molecule,it has much stronger emission capability after excitation.Therefore,although the molecular exciton is initially excited and the population of the NCP is always much smaller than the exciton,the photon flux emitted by the NCP is significantly larger than that directly from the molecule,as depicted in FIG.1(c).In other words,it is mainly the dramatic difference in the radiative decay rates between the NCP and the molecule that determines their respective contributions and the total emission behavior.FIG.1(d)shows the calculated emission spectra of the coupled system by artificially decomposing into the contribution from the NCP and the molecule.As addressed above,due to its weak emission ability of the molecule with respect to the NCP,the emission directly from a single molecule is negligibly small.The total emission spectrum is almost exactly the same as that from the NCP alone,suggesting an overwhelming contribution from the emission mediated by the NCP.Essentially,during the whole emission process,the NCP serves as an antenna,which first accelerates the de-excitation of the molecule by the Purcell effect,speeds up the energy transfer from the molecule to the NCP,and then scatters the induced emission from the molecule to the far field.Based on this and many previous studies[16,19],the scattering process described above will most likely go through an elastic scattering,and thus provide a spectrum with a similar line-shape as the emission directly from the molecule.In other words,the role of the NCP in the situation of initial molecular excitation directly by tunneling electrons is to enhance the molecular emission through the increase of the apparent radiative decay rate,leading to the observation of sharp molecule-specific emission peaks.

FIG.2 (a)Schematic of STM induced NCP emission with the STM tip apex located in close proximity to the edge of a molecule.The molecule cannot be directly excited by tunneling electrons.Instead,the localized NCP in the junction can be excited by inelastic tunneling electrons.The time evolution of the populations(b)and emission intensities(c)of the NCP and the molecule after the NCP is initially excited.(d)Emission spectra for the molecule and the NCP when the NCP is selectively excited.The emission energy for both the molecule and the NCP is set to 2 eV.
On the other hand,when the tip is located in close proximity to the edge of a molecule and only the NCP can be directly excited by the tunneling currents,as schematically illustrated in FIG.2(a),the molecule can be indirectly excited due to the dipolar coupling between the molecule and the NCP.Such coupling could modify both the time evolution and the optical response of the plasmon emission compared with that from a molecule-free tunnel junction.As shown in FIG.2(b),the population of the molecular exciton Pmol(t),which is 0 at t=0,quickly increases and reaches a maximum of∼0.02 at t≈30 fs,and then decays monotonically with time.This behavior can be explained in a similar way to the population of NCP in FIG.1(b):as time increases,the energy in the NCP is rapidly lost and therefore it cannot compensate for the dissipation of the molecular exciton near a plasmonic nanocavity.However,in contrast with the case shown in FIG.1,here the energy loss of the NCP is so fast that the energy of the molecular exciton will conversely compen-sate for the dissipation of the NCP.This can be seen from the population of the NCP PNCP(t)which quickly decreases to 10−7at t≈46 fs and revives to 3.5×10−4at t≈76 fs due to the energy transferred back from the molecule.Indeed,if without dissipation the energy will be exchanged periodically between the exciton and the NCP.As shown in FIG.2(c),due to the weak emission capability of a molecule,the light emission from the single molecule after absorbing the energy from the NCP is negligibly small compared with the emission directly from the NCP.Nevertheless,the single molecule does affect the emission properties of the NCP.Since the linewidth of NCP is usually much larger than that of the molecular exciton,the interaction between the exciton and the NCP can be considered as a coherent dipolar coupling between a single discrete mode and a broader continuous mode,which may result in the occurrence of Fano resonance[13,14,20].When the energy is transferred from the NCP to the molecule and back to the NCP again,it may gain additional phase and cause destructive interferences,manifested by a dip in the emission spectrum,as shown in FIG.2(d).It is worthwhile to note that the dip cannot be simply considered as an incoherent absorption of the NCP emission by the single molecule,since the absorption capability(as its emission capability)is so small that this effect cannot be observed otherwise.Again,the total emission spectrum of the coupled system is essentially the same as the NCP spectrum with Fano dips,and the emission directly from the single molecule is negligibly small.The Fano dip shown in FIG.2(d)is symmetric for the exact resonant condition between the molecule and NCP,which is different from the asymmetric Fano lineshape observed experimentally[13].Such discrepancy between the experimental observation and the current model may be attributed to either the symmetric Lorentzian profiles used to describe the molecule and the NCP or the simple dipolar approximation used to model the molecule-NCP coupling,or both[13].Nevertheless,it should be mentioned that even under the dipolar approximation,when the detuning between the molecule and the NCP is non-zero(i.e.,non-resonant),the Fano lineshape superimposed on the plasmon emission spectrum will become asymmetric according to our model,which is in agreement with experimental observations.
We would also like to note that,according to our theoretical model,while molecule-specific emission peaks will usually be observed when the tip is placed on a well-decoupled molecular emitter due to the strong plasmonic enhancement in the radiative decay rate,the appearance of a Fano-dip like spectrum is still possible if the molecule is non-emissive because the destructive interference between the plasmon and molecule may become dominant and will not be buried by the otherwise enhanced emission peak in this case.On the other hand,when the tip is placed in close proximity to the edge of a molecule and only the NCP is initially excited,we will most probably only see Fano dips superimposed on a broad NCP emission spectrum;it is unlikely to observe molecule-characteristic peaks unless the radiative decay rate of the molecule could become comparable to that of the NCP,which is impossible in reality for a single molecule.
IV.CONCLUSION
In summary,we have theoretically studied the time evolution and spectroscopic properties of the light emission from a coupled plasmon-molecule system when the molecule or the NCP can be selectively excited by tunneling electrons in STM junctions.It has been found that when the molecule is selectively excited,the NCP can enormously enhance the molecular emission via enhancing the apparent radiative decay rate of the molecule.On the other hand,when the NCP is selectively excited,the destructive interferences between the NCP and the single emitter generate Fano dips in the NCP emission spectra.Since in reality the intrinsic radiative decay rate of the NCP is orders of magnitude larger than that of a single emitter,it is unlikely to see sharp molecule-characteristic emission peaks if the molecule is excited indirectly through the nanocavity plasmons.The theoretical model presented in this work provides insight into the microscopic mechanism of plasmon-molecule coupling at the nanoscale and helps to understand and predict the light emission properties from such systems.
V.ACKNOWLE DGMENTS
This work was supported by the National Natural Science Foundation of China,the National Basic Research Program of China,Chinese Academy of Sciences,Anhui Initiative in Quantum Information Technologies,and Basic Research Program of Shenzhen(JCYJ20150401145529035).
[1]X.H.Qiu,G.V.Nazin,and W.Ho,Science 299,542(2003).
[2]Y.Zhang,Y.Luo,Y.Zhang,Y.J.Yu,Y.M.Kuang,L.Zhang,Q.S.Meng,Y.Luo,J.L.Yang,Z.C.Dong,and J.G.Hou,Nature 531,623(2016).
[3]K.Kuhnke,C.Grosse,P.Merino,and K.Kern,Chem.Rev.117,5174(2017).
[4]Z.C.Dong,X.L.Zhang,H.Y.Gao,Y.Luo,C.Zhang,L.G.Chen,R.Zhang,X.Tao,Y.Zhang,J.L.Yang,and J.G.Hou,Nature Photon.4,50(2009).
[5]G.Tian,J.C.Liu,and Y.Luo,Phys.Rev.Lett.106,177401(2011).
[6]Y.Zhang,Y.Zelinskyy,and V.May,Phys.Rev.B 88,155426(2013).
[7]G.Chen,X.G.Li,Z.Y.Zhang,and Z.C.Dong,Nanoscale 7,2442(2015).
[8]G.Chen,X.G.Li,and Z.C.Dong,Chin.J.Chem.Phys.28,552(2015).
[9]M.C.Chong,L.Sosa-Vargas,H.Bulou,A.Boeglin,F.Scheurer,F.Mathevet,and G.Schull,Nano Lett.16,6480(2016).
[10]Y.M.Kuang,Y.J.Yu,Y.Luo,J.Z.Zhu,Y.Liao,Y.Zhang,and Z.C.Dong,Chin.J.Chem.Phys.29,157(2016).
[11]Z.C.Dong,X.L.Guo,A.S.Trifonov,P.S.Dorozhkin,K.Miki,K.Kimura,S.Yokoyama,and S.Mashiko,Phys.Rev.Lett.92,086801(2004).
[12]M.C.Chong,G.Reecht,H.Bulou,A.Boeglin,F.Scheurer,F.Mathevet,and G.Schull,Phys.Rev.Lett.116,036802(2016).
[13]Y.Zhang,Q.S.Meng,L.Zhang,Y.Luo,Y.J.Yu,B.Yang,Y.Zhang,R.Esteban,J.Aizpurua,Y.Luo,J.L.Yang,Z.C.Dong,and J.G.Hou,Nat.Commun.8,15225(2017).
[14]H.Imada,K.Miwa,M.Imai-Imada,S.Kawahara,K.Kimura,and Y.Kim,Phys.Rev.Lett.119,013901(2017).
[15]J.Johansson,P.Nation,and F.Nori,Comput.Phys.Commun.183,1760(2012).
[16]J.R.Lakowicz,Anal.Biochem.337,171(2005).
[17]M.O.Scully and M.S.Zubairy,Quantum Optics,Cambridge,England:Cambridge University Press,(1997).
[18]B.N.Persson and A.Barato ff,Phys.Rev.Lett.68,3224(1992).
[19]K.Aslan,J.R.Lakowicz,and C.D.Geddes,Curr.Opin.Chem.Biol.9,538(2005).
[20]A.E.Miroshnichenko,S.Flach,and Y.S.Kivshar,Rev.Mod.Phys.82,2257(2010).
 CHINESE JOURNAL OF CHEMICAL PHYSICS2018年3期
CHINESE JOURNAL OF CHEMICAL PHYSICS2018年3期
- CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Network Modeling of Inflammatory Dynamics Induced by Biomass Smoke Leading to Chronic Obstructive Pulmonary Disease
- A Double Network Hydrogel with High Mechanical Strength and Shape Memory Properties
- A High-Performance and Flexible Chemical Structure&Data Search Engine Built on CouchDB&ElasticSearch
- Nucleation of Boron-Nitrogen on Transition Metal Surface:A First-Principles Investigation
- Maximum Thermodynamic Electrical Efficiency of Fuel Cell System and Results for Hydrogen,Methane,and Propane Fuels
- Electronic Structure and Optical Properties of K2Ti6O13Doped with Transition Metal Fe or Ag