
离子强度对可变电荷表面吸附性铜离子解吸的影响:高岭石*
2018-06-25 09:09邹献中谢卓文艾绍英
土壤学报 2018年3期
邹献中 陈 勇 谢卓文 艾绍英†
(1 广东省农业科学院农业资源与环境研究所,农业部南方植物营养与肥料重点实验室,广东省养分资源循环利用与耕地保育重点实验室,广州 510640)
(2 佛山市高明区农业技术服务推广中心,广东佛山 528500)
关于可变电荷土壤颗粒表面对重金属离子的吸附机理,已有大量的研究试图解释。作为土壤环境的基本要素,离子强度对可变电荷土壤中重金属离子吸附影响的研究也有相当多的报道[1-5]。然而实际上解吸与环境中相关元素的生物有效性和迁移性更为紧密相关,但关于重金属离子解吸的研究却少得多[6]。Boden等[7]认为,随着被吸附离子浓度的增加,其吸附位置将逐渐远离Stern层,从吸附自由能的角度出发,可以认为,被吸附离子是按照吸附自由能的大小而分层被吸附剂表面吸持。使用惰性电解质进行连续性解吸有助于了解吸附自由能的大小,因为被吸附离子自由能越大,就越容易被低浓度电解质所解吸。
在以往的有关重金属离子的解吸研究中,研究者通常采用各种浓度和不同种类解吸液对吸附性重金属离子进行解吸[8-10],对于能被解吸下来的重金属离子,其原因也通常归结于交换作用[11]。但已有的一些研究结果证明,对于可变电荷土壤和氧化铁,吸附性铜离子也可以在去离子水中解吸,且解吸率可能更大,这一现象的原因已不能归之于交换作用,而更可能是因为离子强度变化导致的表面电位的变化的缘故[12]。但有关这部分可在去离子水中解吸的铜离子与能被交换作用解吸下来的铜离子之间的关系究竟如何所知不多。这一现象是否为氧化物(氧化铁)所特有,还是对可变电荷表面而言普遍存在,还未有定论。由于专性吸附作用的后果之一乃氢离子释放[13-14],因此即使采用初始pH一致的不同重金属离子浓度溶液,氢离子的释放将导致体系最终平衡液pH的不一致性[14]。由于有关反应初始和终了阶段的重金属离子吸附体系pH相关数据的缺失,使得相关研究过程几乎无法完整考虑。联系到pH对重金属离子吸附的显著影响,作为结果,对吸附机理的了解可能是不完全的。
有关连续性解吸对可变电荷土壤和高岭石体系pH的影响的研究结果表明,连续解吸过程中,不同的离子强度变化方向将导致体系pH的变化完全不同,且开始变化的起始点与高岭石ZPC有着密切关系[15]。因为重金属离子同样被可变电荷表面专性吸附,有理由推断离子强度对可变电荷表面吸附重金属离子,比如铜离子,将产生类似的影响。
本研究以高岭石为代表,在单一铜离子浓度条件下,利用去离子水和浓度从低到高的惰性电解质NaNO3对在去离子水以及0.1 mol L-1NaNO3中吸附的Cu(II)进行连续性提取,研究了离子强度变化对高岭石中吸附性Cu(II) 在不同pH段解吸的影响,以更好地了解可变电荷表面对Cu(II)的吸附-解吸机理。
1 材料与方法
1.1 供试材料
供试样品采用德国产Fluka品牌的高岭石,使用电渗析方法对所吸附的杂质离子(主要为钾和钙)进行去除。
1.2 研究方法
铜离子吸附试验:称样品1.000 g 于已知重量的酸洗过的100 ml离心管中,加入已用不同体积0.01 mol L-1NaOH或0.01 mol L-1HNO3溶液调节过pH的去离子水或0.1 mol L-1NaNO3溶液18 ml,所加入酸碱量已经过预实验,以保证最终吸附实验完成时,平衡液pH范围在3.0~6.5之间,且样点分布间隔合适。使用包裹聚四氟乙烯的小磁铁搅拌子和小型搅拌仪充分混合悬液。放置平衡12 h后用微量加液器加入2 ml 0.01 mol L-1Cu(NO3)2标准溶液,恒温振荡箱振荡4 h(25℃,220 r min-1)。吸附平衡后,以3 200 g左右离心力离心4 min,倾出上清液,一部分用玻璃电极法测定pH,一部分用原子光谱吸收法(AAS)测定铜离子浓度。从吸附前后溶液中铜离子的浓度差计算吸附量和吸附率。样品2次重复,吸附率较低部分重复3次。
铜离子连续解吸试验:将上述含残渣的离心管称重,计算土样中残留液的体积,由残留液体积和铜离子浓度,计算残留量。解吸液为去离子水和不同浓度的NaNO3溶液,均用适量0.01 mol L-1NaOH或0.01 mol L-1HNO3溶液调节pH,使其初始pH与将被解吸的样品上次吸附或解吸完成时的平衡液pH基本一致。离心管中加入去离子水,使最终溶液体积为20 ml,搅拌离心等过程同吸附试验。待该次解吸完成后,重复上述过程。其后用浓度由低至高的NaNO3溶液继续解吸,实验过程同上,直至该浓度NaNO3解吸液条件下,Cu2+最高解吸率低于10%为止。由平衡液体积和测定的Cu2+浓度计算出平衡液中Cu2+总量,减去解吸开始前的Cu2+残留量,得到该样次Cu2+解吸量。解吸液浓度和解吸次数依次分别为去离子水(3次)、0.01 mol L-1NaNO3(3次)、0.1 mol L-1NaNO3(3次)和1 mol L-1NaNO3(2次)。
1.3 数据处理
数据处理采用Excel 2010,图中数据均为实测值,作图软件为SigmaPlot13.0。
2 结 果
2.1 pH对高岭石吸附铜离子的影响
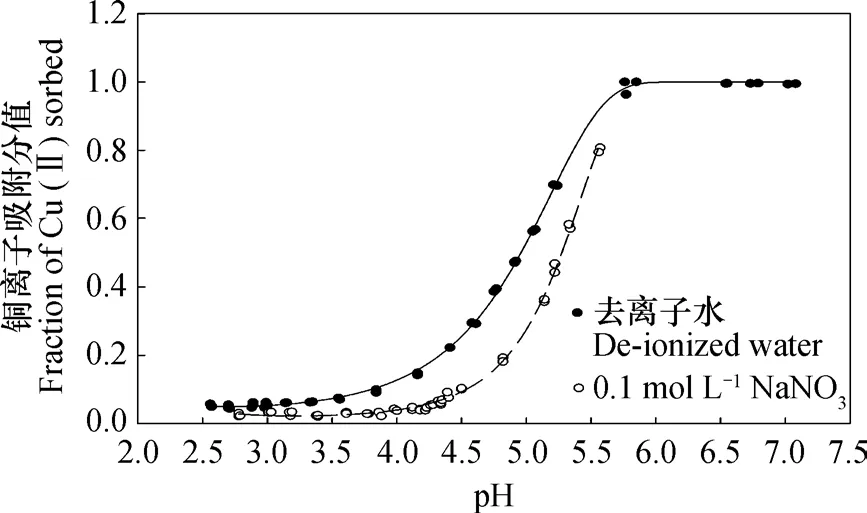
图1 pH对高岭石在去离子水中或0.1 mol L-1 NaNO3溶液中吸附铜离子的影响Fig. 1 Effect of pH on adsorption of Cu(Ⅱ) on kaolinite in de-ionized water or a 0.1 mol L-1 NaNO3 solution
图1是高岭石在去离子水和0.1 mol L-1NaNO3溶液中铜离子吸附分值随pH的变化图。对实测值使用Frisch方程进行拟合,S=1-exp (-(pH/pHe)a),其中a=b+c×pH[16]。拟合方程参数和相关性质见表1。在本试验研究的pH范围(pH2.5~7.3)内,高岭石对Cu2+离子的吸附分值从不足0.1上升至将近1。无论支持电解质浓度大小如何,其pH-Cu(II)吸附分值曲线均可用Frisch方程拟合,且拟合率可达到0.999以上。值得一提的是,无论是在去离子水中或0.1 mol L-1NaNO3溶液中吸附,均在平衡液pH3.7~4.3左右出现拐点。由于Na+对Cu2+的抑制作用,在相同pH条件下,0.1 mol L-1NaNO3溶液中的Cu2+吸附分值要较去离子水中的低。

表1 Frisch方程拟合参数Table 1 Fitting parameters of Frisch Equation
2.2 吸附性铜离子在去离子水中的连续解吸
高岭石吸附铜离子后,在去离子水中解吸时,解吸分值随pH的变化情况如图2所示。实验结果表明,第一,无论吸附时溶液离子强度大或者小,在低pH段,被吸附的铜离子均可在去离子水中解吸,且在相同pH条件下,解吸分值随着解吸次数的增加而减少。第二,在大部分情况下,对于同一种样品,解吸值基本为零时所对应的平衡液pH基本相同,且均为pH5.0左右。第三,用去离子水第一次解吸时,当平衡液pH大于一定值之后,会出现重吸附现象,即当平衡液pH大于一定值之后,Cu2+离子不仅不能被解吸,反而出现了溶液中残余铜离子被高岭石吸附的现象。无论吸附时溶液为去离子水或0.1 mol L-1NaNO3,当第一次用去离子水解吸时,这一现象均可出现。但当吸附液为0.1 mol L-1NaNO3时,相比吸附液为去离子水,其解吸分值的绝对值更高。实验结果同时表明,无论吸附时支持电解质浓度大小,对于第二次和第三次解吸,铜离子解吸分值随pH的上升下降趋势不明显,甚至有先上升后下降的趋势。
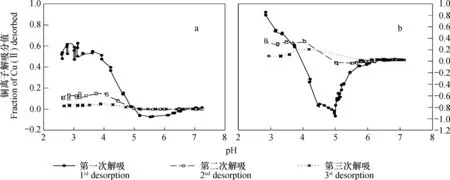
图2 去离子水中解吸时,铜离子解吸分值随pH升高的变化(a:去离子水中吸附;b:0.1 mol L-1 NaNO3 溶液中吸附)Fig. 2 Variation of the fraction of desorbed Cu(II) with increasing pH in de-ionized water(a: adsorbed in de-ionized water; b:adsorbed in 0.1 mol L-1 NaNO3)
2.3 吸附性铜离子在NaNO3溶液中的连续解吸
当样品在去离子水中解吸三次之后,再依次用浓度从低到高的NaNO3溶液解吸时,铜离子解吸分值随pH的变化情况如图3和图4所示。实验结果表明,第一,吸附性铜离子解吸分值随平衡液pH上升的变化规律,并不是单调下降的趋势,而是显现为先上升后下降的峰型。第二,在同一浓度NaNO3条件下,相同pH条件下的解吸分值大小随解吸次数的增加而减小。第三,虽然解吸次数和吸附液NaNO3浓度不同时,在同一浓度NaNO3解吸液和相同解吸平衡液pH条件下,其解吸分值均有不同,但在其他条件,如吸附时支持电解质浓度、解吸次数和解吸平衡液pH,相同时,解吸分值大小随着解吸液支持电解质浓度的增大而依次降低。第四,无论吸附时支持电解质浓度大小如何,在解吸分值急剧增加之前,部分样品的解吸曲线均有一个相对比较平缓的上升过程。
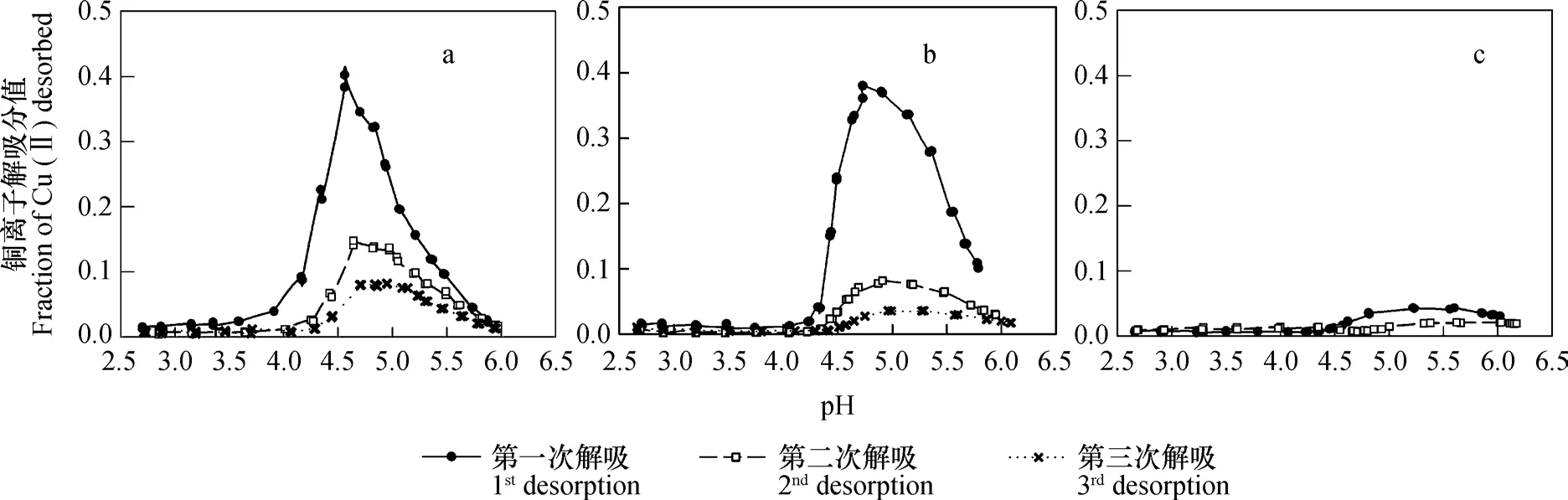
图3 去离子水中吸附后,不同浓度NaNO3溶液中解吸时,铜离子解吸分值随溶液pH升高的变化(a: 0.01 mol L-1 NaNO3 ; b: 0.1 mol L-1 NaNO3; c: 1 mol L-1 NaNO3)Fig. 3 Variation of the fraction of Cu(II)pre-adsorbed in de-ionized water desorbed by NaNO3 with increasing pH and concentration of NaNO3(a: desorbed with 0.01 mol L-1 NaNO3; b: desorbed with 0.1 mol L-1 NaNO3; c: desorbed with 1 mol L-1 NaNO3)
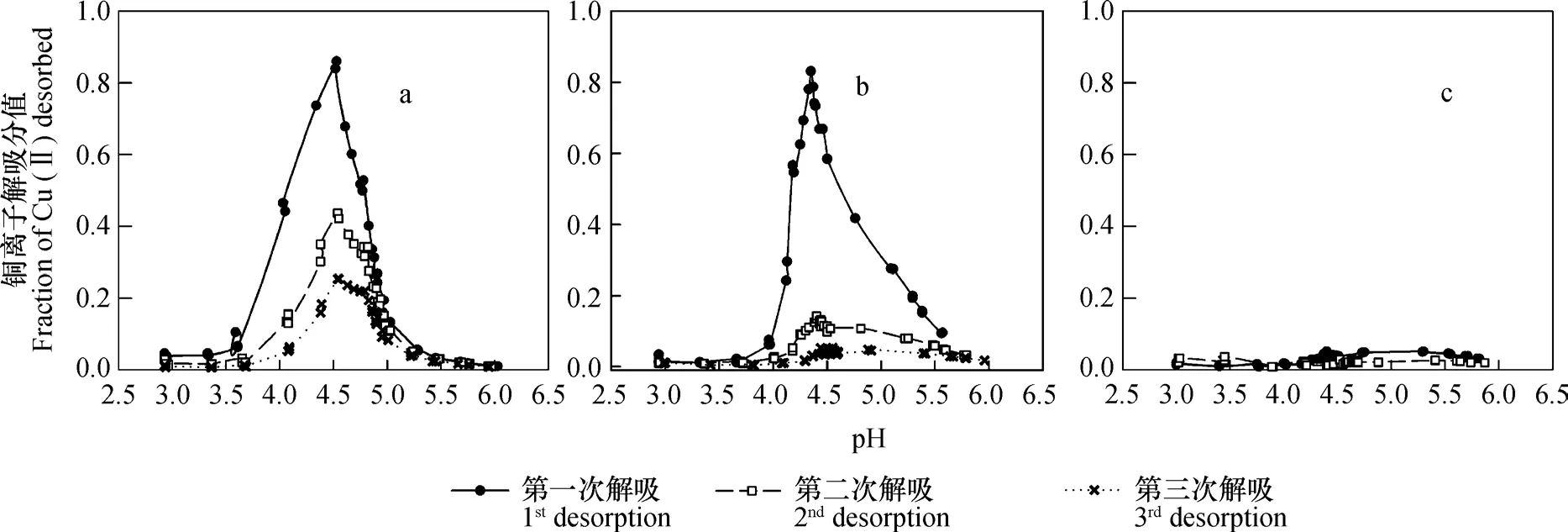
图4 0.1 mol L-1 NaNO3溶液中吸附后,不同浓度NaNO3溶液中解吸时,铜离子解吸分值随溶液pH升高的变化(a: 0.01 mol L-1 NaNO3 ; b: 0.1 mol L-1 NaNO3 ; c: 1 mol L-1 NaNO3)Fig. 4 Variation of the fraction of Cu(II) pre-adsorbed in de-ionized water desorbed by NaNO3 with increasing pH and concentrations of NaNO3 (a: desorbed with 0.01 mol L-1 NaNO3; b: desorbed with 0.1 mol L-1 NaNO3; c: desorbed with 1 mol L-1 NaNO3)
从图中结果可以还看出,当用不同浓度NaNO3溶液进行解吸时,无论解吸次数,在大部分情况下,解吸曲线均可出现解吸分值开始急剧增加的拐点,由于在解吸过程中,体系pH并非不变,因此各拐点的解吸平衡液pH并非一致,但各拐点所对应的pH吸附却基本一致(表2)。对于在去离子水中吸附铜离子者,pH吸附全部均为3.56;对于在0.1 mol L-1NaNO3溶液吸附铜离子者,0.01 mol L-1NaNO3溶液解吸时,均为3.18,0.1 mol L-1NaNO3溶液解吸时,均为3.39。对于同一浓度NaNO3解吸液,pH吸附与解吸次数无关。
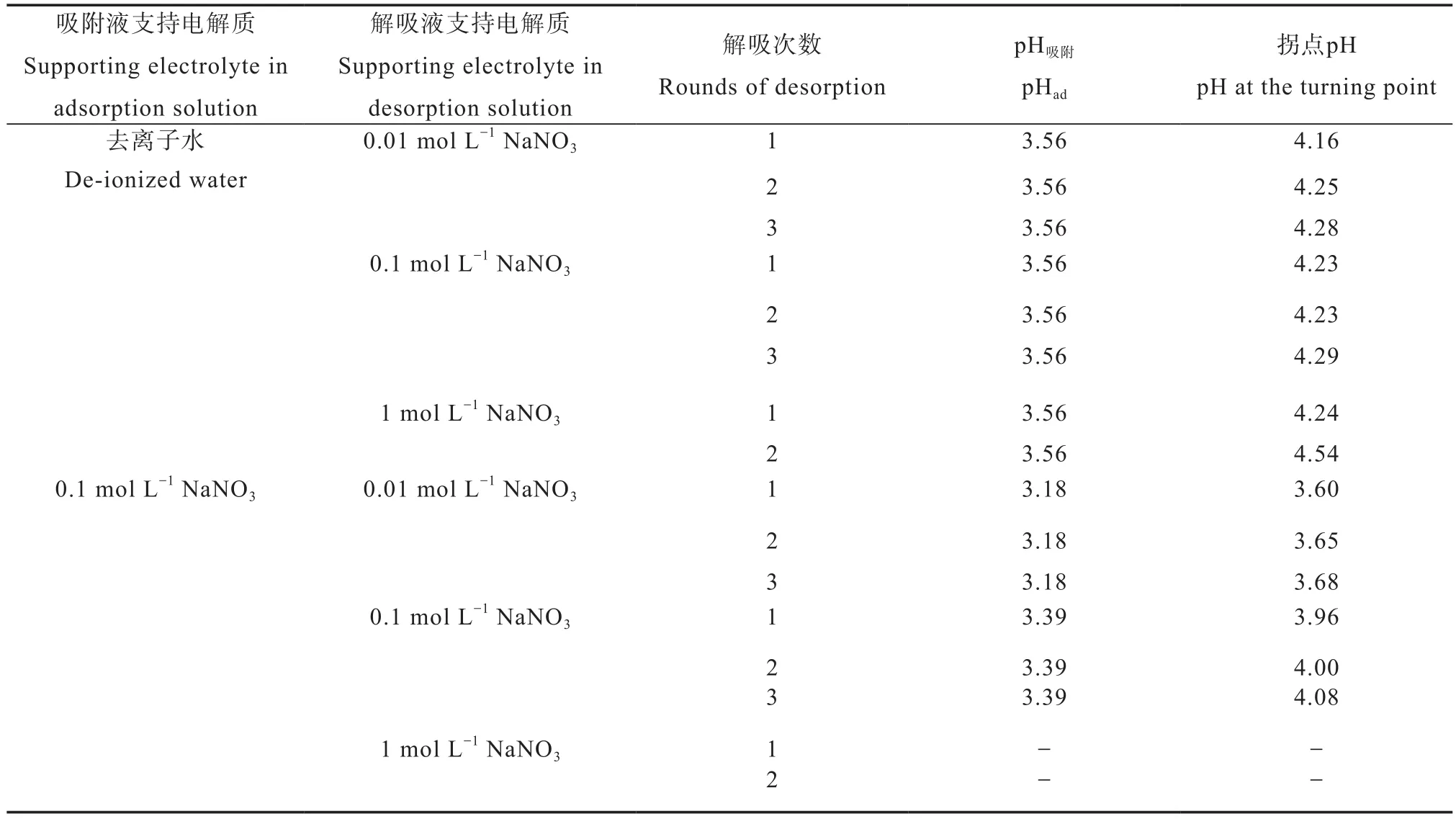
表2 各条件下拐点pH和对应pH吸附Table 2 pH at the turning point and its corresponding pHad relative to desorption condition
3 讨 论
众所周知,土壤或氧化物对重金属离子的吸附包括电性吸附和专性吸附两种,其中由于专性吸附常发生在吸附表面电荷为正电荷的情况下,因此专性吸附通常被认为是因为导致专性吸附的因子的作用强度超过了电性排斥力的结果[17],从这个角度而言,如果电性排斥力增加,可能会导致原本被吸附的离子解吸。由于永久电荷,通常是永久负电荷的电荷量不会因环境因素,如pH等的改变而改变,因此上述电性排斥力增加的情况只可能发生在可变电荷表面,而不会发生在永久电荷表面,已有的实验结果也已经证实了这一点[18]。
可以认为,影响可变电荷表面对离子的专性吸附有两个因素,一个是电荷符号,另一个是离子和吸附表面之间的亲和势和电性排斥(亲和)力的差值(当吸附表面电荷符号与被吸附离子同号时)或加值(当吸附表面电荷符号与被吸附离子异号时)。首先,考虑表面电荷符号的影响。对于某一特定离子,例如,氢离子,如果表面电荷符号由负变正,那么就可能由吸附变成解吸。随着pH的改变,可变电荷表面吸附位电荷符号将发生改变。因此,在不同pH段,吸附或解吸的趋势将发生变化。这就是为什么盐效应零点(PZSE)(pH0)与特征pH(pH特征)密切相关[7,19]。其次,如上所述,当电性排斥力变强时,与吸附表面的亲和势将变弱。根据Boden等[7]提出的四层模型理论,当离子强度减小,吸附表面的表面电位绝对值将增加,因此,离子强度减小的差值越大,电性引力(或斥力)也就越大,这就意味着解吸前后离子强度减小的差值变大将导致对专性吸附离子更强的吸附或解吸(导致吸附还是解吸取决于专性吸附离子和吸附表面的电荷符号是异号还是同号)。罗文贱等[15]用这一理论合理地解释了离子强度变化对可变电荷土壤和高岭石体系pH变化的影响,他们同时明确肯定了离子强度的变化是导致ΔpH变化的主要原因,且与pH特征值密切相关。
需要说明的是,从电性吸附的角度出发,可交换态铜离子可以分为两种亚型:一种是由于黏土矿物中,如高岭石,永久负电荷所引起(永久负电荷型),另一种则是由于高岭石破边面或氧化物表面的可变负电荷所导致(可变电荷型)。
当高岭石表面预先吸附铜离子后,用离子水解吸时,基于上述基本原理,以及由于去离子水中并无其他可交换态离子的存在,同时解吸时去离子水pH与吸附平衡液pH一致,因此,此时解吸或吸附的铜离子均属于可变电荷型。高岭石pH0相对较低(约3.6),对于本研究中所用高岭石样品,电位滴定法测定的结果类似(图5),所以在本文所研究的pH范围内,低pH段时,高岭石表面的可变电荷净电荷应为正电荷,而在其他pH段则为负电荷。根据上述原理,这两部分带不同符号可变电荷表面所吸附的可变电荷型铜离子,在去离子水中解吸时,应该有着完全不同的表现,当pH吸附低于pH特征时,高岭石表面带净可变正电荷,如果离子强度降低,将导致吸附表面和被吸附铜离子之间亲和力的降低,这就会使得铜离子趋向于解吸;反之,将趋向于吸附;当pH吸附高于pH特征时,情况则刚好相反。
以上只是考虑了高岭石表面电荷和离子强度变化对吸附性铜离子的影响,事实上,由于重金属离子吸附是一个连续性过程,且pH变化可能同样影响铜离子的吸附或解吸,因此出现重吸附现象的原因,可以归之以下三点:第一,离子强度减小;第二,体系pH升高;第三,连续性吸附。鉴于相关实验结果已经否认了在本实验条件下的第三种可能性的存在(图6),因此,导致重吸附现象产生的原因只可能是第一或第二种原因。
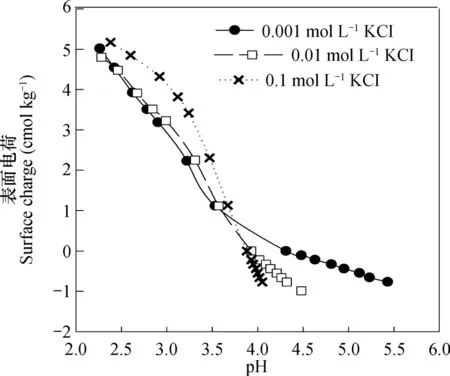
图5 利用电位滴定法测定的高岭石pH0Fig. 5 pH0 of kaolinite determined with potentiometric titration
在去离子水中吸附的铜离子,第一次在去离子水中解吸时,当pH吸附在4.5~6.0时,体系pH的变化ΔpH有异常升高的现象[15],这可能是导致其出现重吸附现象的原因之一。图2中的结果表明,相比在去离子水中吸附铜离子者,在0.1 mol L-1NaNO3溶液中吸附铜离子,然后在去离子水中解吸时,重吸附现象更为明显。这应该是由于相比在去离子水中吸附铜离子,然后用去离子水解吸者,在0.1mol L-1NaNO3溶液中者解吸前后离子强度差更大,因此解吸时的吸附势更强的缘故。这一推论可以通过未电析的高岭石样品在0.1 mol L-1NaNO3溶液中吸附铜离子后再在去离子水中解吸的结果(图7)得到证实。该条件下,在所研究的pH范围内,铜离子的解吸量均为负值,或者说,在所研究的pH范围内,均出现铜离子的重吸附现象。这一实验结果表明,离子强度降低是导致重吸附现象出现的主要原因,因为相比电析后的样品,未电析高岭石中含有大量钾钙离子,这意味着,相比同等条件下已电析的高岭石,其初始离子强度应更大,解吸前后离子强度差值也应更大。
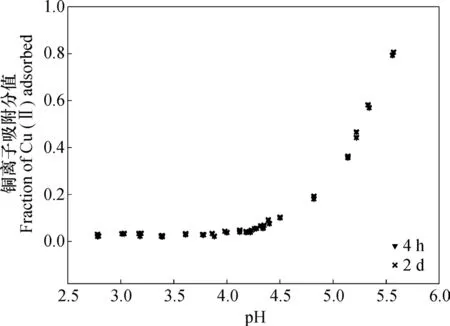
图6 pH和时间(4 h和2 d)对高岭石吸附铜离子的影响Fig. 6 Effect of pH and duration (4 h, 2 d) on adsorption of Cu(II) on kaolinite
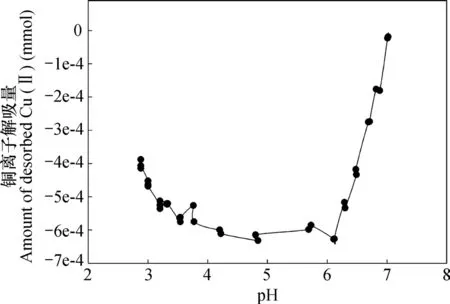
图7 pH对未电析高岭石在0.1 mol L-1 NaNO3溶液中吸附铜离子、去离子水中铜离子解吸量的影响Fig. 7 Effect of pH on adsorption of Cu(II) by unelectrodialyzed kaolinite in 0.1 mol L-1 NaNO3 and desorption of Cu(II) from unelectrodialyzed kaolinite
本研究中,无论解吸次数或者解吸NaNO3浓度大小如何,吸附性铜离子在对应的pH吸附大于pH特征之后,解吸分值均会随着pH的上升而增加,在达到一个峰值后开始下降,这一现象可以用水解作用和高岭石表面可变负电荷数量和密度随pH升高而增加的综合作用解释[5]。
根据上述离子强度变化对吸附性铜离子的影响机理,可以推断,离子强度的增加,应该导致可变电荷表面带净负电荷部分的吸附性铜离子解吸分值的增加,这也应该是为何随着NaNO3浓度的增加,已不能被低浓度NaNO3解吸的铜离子可以在高浓度NaNO3溶液中解吸的原因之一。
图3和图4以及表2的结果表明,解吸分值开始急剧增加时对应的pH解吸在不同解吸条件下并不相同,但与其对应的pH吸附具有高度同一性,这应该是由于当pH吸附低于pH特征时,基本只存在永久负电荷型吸附性铜离子,而这些吸附点或吸附位的表面电位基本上不受pH或离子强度影响,此时铜离子的解吸应只是交换性钠离子增加的缘故;当pH吸附高于pH特征时,高岭石表面可变净电荷由正转变为负,同时因高岭石的可变电荷深受pH和离子强度的影响,以及高岭石边面的诱导水解作用,使得相对永久负电荷型铜离子,可变电荷型铜离子的解吸分值随pH和离子强度的变化而急剧变化。这也再一次证实,影响吸附性铜离子解吸的一个重要因素是高岭石的pH0,同时也表明,在此段pH,高岭石对铜离子的吸附应主要以可变电荷型为主。
4 结 论
高岭石在去离子水和0.1 mol L-1NaNO3溶液中吸附铜离子后,当依次在去离子水中和浓度由低到高的NaNO3溶液中解吸时,由于解吸液离子强度变化导致的解吸分值随pH的升高的变化实质上是因为离子强度变化对可变电荷表面电位,以及Na+交换作用、pH变化对高岭石边面诱发水解作用和土壤表面电荷性质影响的综合体现,其中高岭石的表面电荷性质对这一变化的影响尤为重要,这也显示了可变电荷表面的重金属离子吸附-解吸机理的复杂性。
[ 1 ] Cao J,Lam K C,Dawson R W,et al. The effect of pH,ion strength and reactant content on the complexation of Cu2+by various natural organic ligands from water and soil in Hong Kong. Chemosphere,2004,54:507—514
[ 2 ] Wang Y,Tang X W,Chen Y M,et al. Adsorption behavior and mechanism of Cd(II) on loess soil from China. Journal of Hazardous Material,2009,172:30—37
[ 3 ] Zhu B,Alva A K. Differential adsorption of trace metals by soils as influenced by exchangeable cations and ionic strength. Soil Science,1993,155:61—66
[ 4 ] 杨少海,陈勇,刘辉,等. 离子强度对铁质砖红壤铜离子连续解吸的影响. 土壤学报,2014,51(6):1290—1297 Yang S H,Chen Y,Liu H,et al. Effect of ionstrength on successive desorption of copper ions in Hyper-Rhodic Ferralsol (In Chinese). Acta Pedologica Sinica,2014,51(6):1290—1297
[ 5 ] Jiang J,Wang Y,Xu R K,et al. Adsorption of chromate on variable charge soils as influenced by ionic strength. Environmental. Earth Science,2012,66:1155—1162
[ 6 ] Paripurnanda L,Saravannamuthu V,Jaya K.Cadmium sorption and desorption in soils:A review.Environmental Science & Technology. 2012,42:489—533
[ 7 ] Boden J W,Nagarajah S, Barrow N J,et al.Describing the adsorption of phosphate,citrate and selenite on a variable charge mineral surface.Australian Journal of Soil Research,1980,18:49—60
[ 8 ] Moreno A M,Quintana J R,Pérez L,et al. Factors influence lead sorption-desorption at variable added metal concentrations in Rhodoxeralfs. Chemosphere,2006,64:758—763
[ 9 ] Schramel O,Michalke B,Kettrup A. Study of the copper distribution in contaminated soils of hop fields by single and sequential extraction procedures. Science of the Total Environmental,2000,263:11—22
[10] Ma L,Xu R K,Jiang J. Adsorption and desorption of Cu(II) and Pb(II) in paddy soils cultivated for various years in the subtropical China. Journal of Environmental Sciences,2010,22:689—695
[11] 季国亮,李洪燕. 阳离子的电性吸附//于天仁,季国亮,丁昌璞,等. 可变电荷土壤的电化学. 北京:科学出版社,1996 Ji G L,Li H Y,Electrostatic adsorption of cations//Yu T R,Ji G L,Ding C P,et al. Electrochemistry of variable charge soils (In Chinese). Beijing:Science Press,1996
[12] 邹献中,张超兰,宁建凤,等. 不同浓度铜离子土壤的吸附-解吸行为——兼论弱专性吸附态的存在. 土壤学报,2012,49(5):892—900 Zou X Z,Zhang C L,Ning J F,et al. Behaviors of copper ions different in concentration in sorptiondesorption by soils—And existence of weak-specificadsorption state (In Chinese). Acta Pedologica Sinica,2012,49(5):892—900
[13] 邹献中,赵安珍,季国亮. 可变电荷土壤吸附铜离子时氢离子的释放. 土壤学报,2002,39(3):308—317 Zou X Z,Zhao A Z,Ji G L. Release of hydrogen ions during adsorption of copper ions by variable charge soils (In Chinese). Acta Pedologica Sinica,2002,39(3):308—317
[14] 邹献中,张超兰,魏岚,等. 不同浓度铜离子土壤的吸附-解吸行为. 土壤学报,2011,48(5):964—970 Zou X Z,Zhang C L,Wei L,et al. Effect of electrolyte concentration on release of hydrogen ions from soils adsorbing copper ions (In Chinese). Acta Pedologica Sinica,2011,48(5):964—970
[15] 罗文贱,张政勤,陈勇,等. 连续解吸中离子强度对可变电荷土壤和高岭石体系pH的影响. 土壤学报,2016,53(1):146—154 Luo W J,Zhang Z Q,Chen Y,et al. The effect of ionic-strength change on the system pH of variable charge soils and kaolinite during successive desorption(In Chinese). Acta Pedologica Sinica,2016,53(1):146—154
[16] Fischer L,Brummer G W, Barrow N J. Observations and modelling of the reactions of 10 metals with goethite:Adsorption and diffusion processes. European Journal of Soil Science,2007,58:1304—1315
[17] 于天仁,孙含元,张宏. 阳离子专性吸附//于天仁,季国亮,丁昌璞,等. 可变电荷土壤的电化学. 北京:科学出版社,1996 Yu T R,Sun H Y,Zhang H,Specific adsorption of cations//Yu T R,Ji G L,Ding C P,et al.Electrochemistry of variable charge soils (In Chinese). Beijing:Science Press,1996
[18] 邹献中,徐建民,赵安珍,等. 可变电荷土壤中铜离子的解吸. 土壤学报,2004,41(1):68—73 Zou X Z,Xu J M,Zhao A Z,et al. Desorption of copper ions adsorbed by variable charge soils (In Chinese). Acta Pedologica Sinica,2004,41(1):68—73
[19] Barrow N J. The four laws of soil chemistry:the Leeper lecture 1998. Australian Journal of Soil Research,1999,37(5):787—830
猜你喜欢
江苏农业科学(2022年19期)2022-10-28
矿产综合利用(2021年5期)2022-01-17
氯碱工业(2021年6期)2021-12-25
中学生数理化·中考版(2021年10期)2021-11-22
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
石油化工应用(2020年7期)2020-08-08
矿产综合利用(2020年1期)2020-07-24
石油地质与工程(2020年3期)2020-06-24
太原理工大学学报(2019年3期)2019-05-30
玻璃纤维(2016年2期)2016-12-18
