
脑缺血再灌注损伤中自由基与锌离子的相互作用
2018-06-23 09:03房亚兰刘克建赵咏梅
首都医科大学学报 2018年3期
房亚兰 尹 洁 刘克建 赵咏梅
(首都医科大学宣武医院 北京市老年病医疗研究中心 神经变性病教育部重点实验室,脑血管病转化医学北京市重点实验室,北京 100053)
脑缺血后再灌注可挽救濒临死亡的细胞,使某些可逆性损伤获得功能上的恢复。但缺血后的血流恢复能进一步导致组织损伤和功能障碍,称为缺血再灌注损伤。降低再灌注损伤被认为是治疗缺血性脑卒中的关键。近年来的研究[1-2]表明,脑缺血再灌注不但诱导大量的自由基产生,引发氧化应激,而且引起细胞内锌离子(Zn2+)大量聚集。本文拟对脑缺血再灌注损伤过程中自由基与Zn2+的相互关系进行阐述。
1 脑缺血再灌注时自由基的产生途径及损伤机制
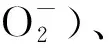
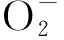
最近的研究[8]表明,再灌注24 h内,在大脑中动脉阻塞(middle cerebral artery occlusion,MCAO)模型大鼠的半暗带区内ROS随再灌注时间延长递增,且ROS量与脑梗死体积比率呈正相关。给予MCAO大鼠线粒体ROS抑制剂,可显著降低大鼠再灌注后6 h脑内ROS的产生,减少脑梗死体积,说明ROS在脑缺血再灌注损伤中发挥重要作用[9]。研究[10]显示,缺血后锥体神经元内自由基的产生与线粒体通透性改变相关。线粒体通透性转运孔(mitochondrial permeability transition pore, mPTP)开放引起促凋亡蛋白释放,例如凋亡诱导因子(apoptosis-inducing factor, AIF)和细胞色素C,后者可激活Caspase-9,进而激活Caspase-3,引起细胞凋亡[10]。因此,脑缺血后,自由基可通过引发细胞凋亡途径参与神经细胞的损伤过程。
2 脑缺血再灌注后脑组织内Zn2+的变化及作用
锌元素是体内必需的微量元素之一,但过量的锌有细胞毒性,导致细胞凋亡。生理条件下,Zn2+和谷氨酸一起储存在神经元的突触囊泡中,受到刺激时,Zn2+释放入突触间隙,作用于突触后的锌结合位点[11]。此外,金属硫蛋白(metallothionein, MT)结合大量的Zn2+,其中MT3对于保持神经元中的锌稳态有重要作用[12]。Zn2+也聚集于线粒体基质中,线粒体内的Zn2+流可通过去极化以及mPTP的开放导致胞质内Zn2+水平增加[13]。除此之外,胞内其他结合位点也可能释放Zn2+,调控缺血后Zn2+水平的病理性上升。研究[2]表明,MCAO大鼠脑梗死半暗带区域内,Zn2+阳性细胞数目在再灌注24 h内递增,且Zn2+阳性细胞数目与脑梗死体积比率呈正相关,说明Zn2+参与脑缺血再灌注损伤。
笔者的研究[2]表明,MCAO大鼠脑内半暗带区Zn2+与凋亡神经元共定位。应用特异性锌离子螯合剂N,N,N′,N′-4(2-吡啶甲基)乙二胺[N,N,N′,N′-tetrakis(2-pyridylmethyl) ethylenediamine, TPEN]显著减少缺血大鼠脑半暗带区Zn2+与凋亡神经元数目,减小脑梗死体积,改善神经功能,且这种保护作用可持续至再灌注后的第14天,首次证明Zn2+在脑缺血损伤中有重要作用[2]。笔者的研究[14]还显示,过量的锌积累于缺血大鼠的微血管,TPEN显著改善缺血大鼠脑内微血管的通透性,说明锌释放和积累导致脑缺血后的血-脑脊液屏障损害。
缺血后Zn2+对神经元线粒体的作用机制十分复杂。Zn2+可抑制线粒体的电子传递,不可逆地阻断重要的线粒体基质酶,进而导致mPTP激活[13]。缺血条件下,内源性Zn2+与线粒体功能紊乱密切相关。锌离子螯合剂可减少缺血起始阶段线粒体细胞色素C释放,抑制Caspase 3激活。亚微摩尔浓度的Zn2+可同时引起线粒体内膜多个离子通道激活以及mPTP开放[15],引起凋亡。
除了影响线粒体功能外,Zn2+也参与调节细胞的其他代谢过程。如在缺血条件下,Zn2+调节AMPA通道、红藻氨酸通道(Ca-A/K通道)GluR2亚基表达,增加Ca-A/K通道对钙离子(Ca2+)的通透性[16]。Zn2+也可以通过激活膜上MAPKs家族介导神经元损伤[17]。然而另有研究[18]表明,Zn2+调控转录因子表达是预适应的保护机制之一。以上关于锌离子在脑缺血再灌注损伤中的相关研究,说明锌离子是一个非常关键的靶点,具有重要的研究意义和广阔的应用前景。
3 Zn2+与自由基引发的氧化应激损伤之间的相关性
Zn2+和ROS在缺氧缺血性应激过程中积累,并在病理过程中起重要作用。Zn2+是一种潜在的有毒阳离子,参与脑缺血、癫痫和脑外伤中的神经元损伤。一方面,Zn2+发挥其神经毒性的机制包括导致线粒体和线粒体外产生活性氧簇,以及破坏代谢酶的活性,最终导致凋亡和/或坏死过程的激活。各种针对Zn2+毒性的神经保护措施能够降低Zn2+引起的ROS生成。另一方面,细胞内Zn2+稳态对病理生理环境变化敏感,如氧化应激、酸中毒或炎性反应。抗氧化剂能够减弱Zn2+的神经毒性,这些都证明Zn2+与氧化应激损伤之间密切相关。
4 脑缺血再灌注过程中Zn2+对氧化应激的调控作用
近年来,脑缺血再灌注后Zn2+对氧化应激的影响受到关注。文献[19-25]报道,Zn2+可促进自由基产生,引起氧化应激损伤。本课题组的研究[19]显示,锌超载导致缺血神经元线粒体功能障碍。细胞内升高的Zn2+可通过线粒体内级联反应促使ROS生成。有研究[20]表明,脑缺血时线粒体对Zn2+的摄取增加,线粒体内快速大量聚集的Zn2+通过抑制电子传递链的复合体Ⅲ的激活或干扰复合体Ⅰ和α-酮戊二酸脱氢酶促使大量的ROS产生,ROS能够引起线粒体膜电位变化,使线粒体功能丧失。尽管Zn2+与Ca2+都可引起上述变化,然而与Ca2+相比,由Zn2+介导的线粒体功能失调的作用更为强烈[20]。
Zn2+升高后也可通过非线粒体途径引发Zn2+依赖性的氧化应激反应。在培养的神经元中,Zn2+可以激活蛋白激酶C(protein kinase C, PKC),进而导致NADPH氧化酶激活,促使NOS生成,同时ROS、NO会相应增加[21]。另一种可能方式是通过nNOS介导,由于局灶性脑缺血时nNOS阳性神经元增加,此途径可能参与了脑缺血损伤[22]。
Zn2+通过线粒体损伤或NADPH氧化酶激活引起氧化应激反应,进而促进ROS产生,诱导DNA链断裂,导致DNA修复酶-多聚核糖聚合酶[poly(ADP-ribose)polymerase, PARP]激活。过量的PARP通过以NAD+作底物催化蛋白质的多聚(ADP-核糖基)化,导致NAD+大量消耗、糖代谢障碍、ATP缺失。应用NADPH氧化酶、NOS或PARP的阻断剂均可抑制Zn2+破坏神经元[23]。PARP激活诱导线粒体释放AIF,促进AIF转入核内,引起非Caspase依赖的DNA断裂及细胞死亡。此外,PAR(PARP的多聚体产物)可以直接诱导AIF释放[26]。体内研究[2]表明,脑缺血导致PARP-1的裂解和激活,而螯合Zn2+后PARP-1的裂解和激活都降低,说明Zn2+参与PARP-1的裂解和激活,促进神经细胞死亡。
Zn2+也通过溶酶体介导细胞死亡。有研究[27]显示,暴露在H2O2或毒性水平的Zn2+环境时,溶酶体内锌迅速增加,导致膜的完整性破坏,溶酶体释放组织蛋白酶,最终海马区神经元通过锌、组织蛋白酶途径死亡。这些结果显示,锌是氧化应激和溶酶体膜通透性连接的纽带,溶酶体锌超载和溶酶体损坏是神经元氧化应激死亡的关键步骤。
尽管一些研究[20,23]表明,Zn2+可促进氧化应激损伤,但另一些报道[28-29]显示,Zn2+具有抗氧化作用。许多含锌蛋白或被锌调节的蛋白能直接或间接影响细胞的氧化平衡。研究[28]表明,细胞氧化应激损伤与Zn2+下降相关联,Zn2+浓度增加可阻止氧化应激损伤。缺少Zn2+的培养神经元和动物模型氧化信号激活,且通过下调细胞内的谷胱甘肽(glutathione, GSH)降低细胞对促氧化应激物质的承受能力[29]。
5 脑缺血再灌注后氧化应激对Zn2+的调控作用
脑缺血时显著发生的氧化应激和酸中毒能够诱导Zn2+从MT蛋白释放,引起细胞内Zn2+增加,这种机制已经在培养神经元中得到证实,并可能是ZnT3敲除动物海马锥体神经元内Zn2+增加的机制[30]。研究[31]显示,大鼠在含氧量正常但血糖低的情况下,可以引发NO应激,最终引起Zn2+聚集。这可能是通过NMDAR介导的nNOS途径实现的。这种NO依赖性的Zn2+增加可以激活NADPH氧化酶及PARP1,诱导细胞死亡。尽管自由基对锌离子的调控作用已逐渐成为研究热点,但目前有关这方面的研究还十分有限。探究氧化应激对锌离子的调控,必将有助于揭示脑缺血损伤的机制。
综上所述,在脑缺血再灌注损伤中,自由基与Zn2+之间存在相互作用,Zn2+可能通过线粒体或NADPH氧化酶等途径促进ROS产生,ROS也可以促进Zn2+从MT等存储位点释放,两者相互作用促进神经损伤。Zn2+在特定条件下又可以发挥抗氧化作用。但迄今为止,Zn2+促进氧化应激并产生损伤的分子机制还没有完全阐明,ROS对Zn2+的调控作用及机制更是知之甚少。因此,脑缺血再灌注损伤中自由基与Zn2+的相互作用值得深入研究,这将为开发脑缺血损伤的保护措施提供新策略。
[1] Taskiran-Sag A, Yemisci M, Gursoy-Ozdemir Y, et al. Improving microcirculatory reperfusion reduces parenchymal oxygen radical formation and provides neuroprotection [J]. Stroke, 2018,49(5):1267-1275.
[2] Zhao Y, Pan R, Li S, et al.Chelating intracellularly accumulated zinc decreased ischemic brain injury through reducing neuronal apoptotic death [J]. Stroke, 2014, 45(4):1139-1147.
[3] Zhang Y, Zhou H, Wu W, et al. Liraglutide protects cardiac microvascular endothelial cells against hypoxia/reoxygenation injury through the suppression of the SR-Ca2+-XO-ROS axis via activation of the GLP-1R/PI3K/Akt/survivin pathways [J]. Free Radic Biol Med, 2016, 95:278-292.
[4] Cadenas S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection [J]. Free Radic Biol Med, 2018, 117:76-89.
[5] Carbone F, Teixeira P C, Braunersreuther V, et al. Pathophysiology and treatments of oxidative injury in ischemic stroke: focus on the phagocytic NADPH oxidase 2 [J]. Antioxid Redox Signal, 2015, 23(5):460-489.
[6] Brennan A M, Suh S W, Won S J, et al. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation [J]. Nat Neurosci, 2009, 12(7):857-863.
[7] AshwaI S, Tone B, Tian H R, et al. Core and penumbral nitric oxide synthase activity during cerebral ischemia and reperfusion [J]. Stroke, 1998, 29(5):1037-1047.
[8] 闫峰, 赵咏梅, 罗玉敏, 等. 大鼠局灶性脑缺血再灌注脑组织中活性氧自由基表达的时程变化[J]. 首都医科大学学报, 2015, 36(5):694-698.
[9] 尹洁, 闫峰, 罗玉敏, 等. R(+)-普拉克索对大鼠短暂性局灶性脑缺血损伤的神经保护作用[J]. 首都医科大学学报, 2014, 35(2):225-230.
[10] Gasche Y, Copin J C, Sugawara T, et al. Matrix metalloproteinase inhibition prevents oxidative stress-associated blood-brain barrier disruption after transient focal cerebral ischemia [J]. J Cereb Blood Flow Metab, 2001, 21(12):1393-1400.
[11] Kawahara M, Tanaka K I, Kato-Negishi M. Zinc, carnosine, and neurodegenerative diseases [J]. Nutrients, 2018, 10(2),Pii:E147.
[12] 闫峰, 刘克建, 赵咏梅, 等. 中枢神经系统中锌离子释放的相关机制及其与脑缺血损伤的关系[J]. 神经疾病与精神卫生, 2014, 14(4):406-409.
[13] Gazaryan I G, Krasinskaya I P, Kristal B S, et al. Zinc irreversibly damages major enzymes of energy production and antioxidant defense prior to mitochondrial permeability transition [J]. J Biol Chem, 2007, 282(33):24373-24380.
[14] Qi Z, Liang J, Pan R, et al. Zinc contributes to acute cerebral ischemia-induced blood-brain barrier disruption [J]. Neurobiol Dis, 2016, 95:12-21.
[15] Wang G, Huang H, Zheng H, et al. Zn2+and mPTP mediate endoplasmic reticulum stress inhibition-induced cardioprotection against myocardial ischemia/reperfusion injury [J]. Biol Trace Elem Res, 2016, 174(1):189-197.
[16] 李森, 刘克建, 赵咏梅, 等. 锌离子在脑缺血中作用的研究进展[J]. 首都医科大学学报, 2013, 34(1):75-79.
[17] Irving E A, Bamford M. Role of mitogen-and stress-activated kinases in ischemic injury [J]. J Cereb Blood Flow Metab, 2002, 22(6):631-647.
[18] Aras M A, Hara H, Hartnett K A, et al. Protein kinase C regulation of neuronal zinc signaling mediates survival during preconditioning [J]. J Neurochem, 2009, 110(1):106-117.
[19] Dong W, Qi Z, Liang J, et al. Reduction of zinc accumulation in mitochondria contributes to decreased cerebral ischemic injury by normobaric hyperoxia treatment in an experimental stroke model [J]. Exp Neurol, 2015, 272:181-189.
[20] Ji S G, Weiss J H. Zn2+-induced disruption of neuronal mitochondrial function: synergism with Ca2+, critical dependence upon cytosolic Zn2+buffering, and contributions to neuronal injury [J]. Exp Neurol, 2018, 302:181-195.
[21] Aimo L, Cherr G N, Oteiza P I,et al. Low extracellular zinc increases neuronal oxidant production through NADPH oxidase and nitric oxide synthase activation [J]. Free Radic Biol Med, 2010, 48(12):1577-1587.
[22] Wang H R, Li J S, Chen J, et al. Effects of zinc on activity of NOS and expression of nNOS in hippocampus of acute hypoxic mice [J]. Zhongguo Ying Yong Sheng Li Xue Za Zhi, 2006, 22(4):395-398.
[23] Shuttleworth C W, Weiss J H. Zinc: new clues to diverse roles in brain ischemia [J]. Trends Pharmacol Sci, 2011, 32(8):480-486.
[24] 曾晓莉, 张文娟,赵博,等.慢性脑缺血神经保护机制及药物研究进展[J].中国脑血管病杂志, 2017,14(11):607-611.
[25] 刘云, 高晓莹,戚思华.缺血后处理脑保护作用的研究进展[J].中国脑血管病杂志, 2015,12(3):160-164.
[26] Andrabi S A, Kim N S, Yu S W, et al. Poly(ADP-ribose) (PAR) polymer is a death signal [J]. Proc Natl Acad Sci U S A, 2006, 103(48):18308-18313.
[27] Hwang J J, Lee S J, Kim T Y, et al. Zinc and 4-hydroxy-2-nonenal mediate lysosomal membrane permeabilization induced by H2O2in cultured hippocampal neurons [J]. J Neurosci, 2008, 28(12):3114-3122.
[28] Oteiza P I. Zinc and the modulation of redox homeostasis [J]. Free Radic Biol Med, 2012, 53(9):1748-1759.
[29] Omata Y, Salvador G A, Supasai S, et al. Decreased zinc availability affects glutathione metabolism in neuronal cells and in the developing brain [J]. Toxicol Sci, 2013, 133(1):90-100.
[30] Lee J Y, Kim J H, Palmiter R D, et al. Zinc released from metallothionein-iii may contribute to hippocampal CA1 and thalamic neuronal death following acute brain injury [J]. Exp Neurol, 2003, 184(1):337-347.
[31] Suh S W, Hamby A M, Gum E T, et al. Sequential release of nitric oxide, zinc, and superoxide in hypoglycemic neuronal death [J]. J Cereb Blood Flow Metab, 2008, 28(10):1697-1706.
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
海洋通报(2021年1期)2021-07-23
生物学通报(2021年4期)2021-03-16
生物化工(2021年2期)2021-01-19
昆明医科大学学报(2020年11期)2020-12-28
生物化工(2020年1期)2020-02-17
读与写(2019年35期)2019-11-05
现代职业教育·高职高专(2018年7期)2018-05-14
中成药(2018年4期)2018-04-26
中国康复理论与实践(2015年10期)2015-12-24
