
二氧化碳直接合成碳酸二甲酯研究进展
2018-05-24 02:23:46李聪明
天然气化工—C1化学与化工 2018年2期
刘 斌,喻 健,李聪明,李 忠
(太原理工大学煤化工研究所,煤科学与技术教育部和山西省重点实验室,太原 030024)
CO2是头号温室气体,同时也是丰富和廉价的碳源,将CO2捕集并转化为高附加值的产品是CO2减排和利用的重要途径。目前,CO2的转化方式有:CO2加氢转化为甲醇或者烃类、CO2和甲烷重整转化为合成气、CO2和醇类或者环氧化物转化为碳酸酯等[1]。
碳酸二甲酯(DMC)是一种用途广泛的绿色有机化工产品,可以代替剧毒的硫酸二甲酯和光气与多种醇、酚、肼类化合物进行反应合成许多高附加值的下游产品,也可以用作环保型的油品添加剂。另外,DMC也是优良的有机溶剂,用于涂料和锂电池等[2]。
DMC的合成方法主要有光气法、尿素醇解法、甲醇氧化羰基化法、CO2与甲醇直接合成法。CO2直接合成法对环境友好,原子利用率高,原料廉价丰富,因此受到越来越多的关注。
1 CO2和甲醇直接合成碳酸二甲酯的反应体系
1.1 CO2超临界反应体系
处于超临界状态的CO2具有良好的溶解特性和传质特性,因此超临界CO2经常被用作有机反应中的溶剂。在CO2和甲醇合成DMC的反应中,CO2处于超临界状态,既做了反应体系的溶剂,又做反应物参与反应。孔令丽等[3,4]利用催化剂Cu2(μ-OEt)2/SiO2在CO2超临界条件下催化合成DMC,发现处于超临界条件下的甲醇转化率(~4.5%)远高于非超临界条件下的甲醇转化率(~2.0%)。林春锦等[5]在CO2超临界条件下,采用CO2、环氧丙烷(PO)和CH3OH为原料,CH3OK为催化剂,无水CaCl2为脱水剂,一步法合成了DMC。研究结果表明催化剂在CO2超临界条件下活性最高,若压力继续升高活性反而下降,在反应温度140℃下,反应3h后DMC的最大收率为48%。
1.2 离子液体反应体系
离子液体是一种性能优良的绿色溶剂,具有较低的熔点,并且离子液体的稳定性较好,近年来越来越广泛地被用做合成反应中的溶剂和催化剂。通常情况下离子液体是由有机阳离子和无机阴离子构成,因此可通过调变不同的阴阳离子组合,设计合成功能不同的离子液体,并且组成离子液体的阴阳离子不同,催化剂表面的酸碱性也会不同。在CO2和甲醇直接合成DMC的反应中,加入离子液体,既可以起到溶剂的作用,又可以对反应起到催化作用。Ding等[6]利用聚乙二醇与BMim系离子液体合成了两种具有不同分子结构的催化剂A和B,离子液体A中聚乙二醇通过C-O键的作用与咪唑环上C=N键中的C原子连接,使C=N双键变为C-N单键,PEG在保持原有的C-C-O骨架结构不变的情况下,咪唑环上原有的甲基和丁基结构不变,而在催化剂B的结构中,PEG通过C-O键作用连接在咪唑环上C=N的C原子上,使得N原子上的丁基脱落,形成仲胺基-NH-结构,而咪唑环上原有的甲基结构保持不变。经过活性评价后,发现催化剂A成功催化合成的单程产率为20.78%,而催化剂B却没有催化活性。这表明催化剂结构中,聚乙二醇通过C-O键与五元共轭环生成的新结构以及连接在共轭环中N原子上的丁基均对催化CO2和甲醇直接合成DMC具有关键作用。
1.3 膜反应器反应体系
在催化反应中使用膜反应器,通过及时分离产品可以促进反应转化,抑制或避免副反应,提高反应选择性和产品的收率。在CO2合成DMC的反应中使用膜反应器可以迅速、持续的将水从反应体系脱除,极大的提高DMC的产率。Zhong等[7]人采用溶胶凝胶法制备了三种不同的膜反应器:二氧化硅无机膜、聚酰亚胺二氧化硅和聚酰亚胺二氧化钛杂化膜,然后将Cu催化剂和膜反应器负载在MgO-SiO2上。经过活性测试后,相比于没有使用膜反应器的体系,使用膜反应器DMC的产率增加了1.54%,2.65%和2.25%,DMC的选择性增加了1.4%,6%和3.3%。虽然提高的程度不太理想,但是加入膜反应器后DMC的产率确实得到了改进,这也为后人的研究方向提供了一个不错的依据。
2 CO2和甲醇直接合成DMC催化剂研究进展
2.1 均相催化剂
2.1.1 金属烷氧基有机化合物催化剂
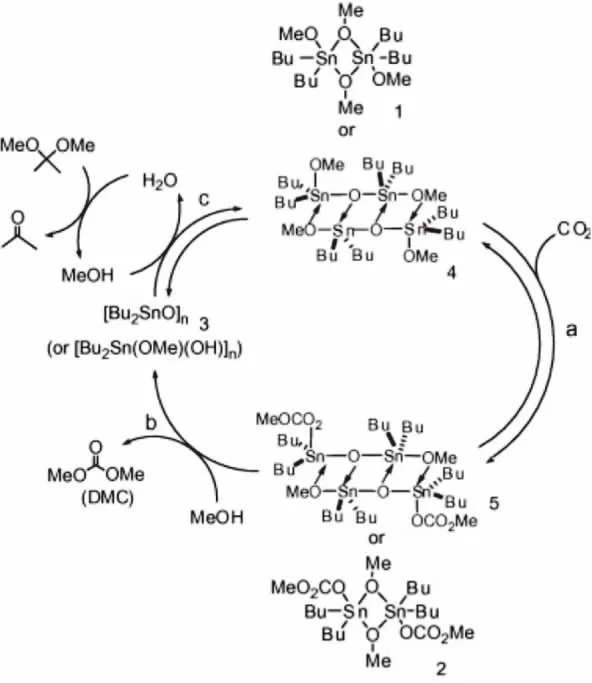
图1 有机锡催化剂催化合成DMC的可能反应机理
有机烷氧基金属催化剂是较早用于催化CO2和甲醇直接合成碳酸二甲酯的催化剂体系,其中具有代表性的催化剂是有机锡类催化剂。Kizlink等[8]考察了一系列含有不同烷基的有机锡催化剂:Bu2Sn(OR)2(OR=Me,Et,Bu),在 130~190℃的反应温度下,反应6h后DMC的收率为160%(以有机锡化合物的物质的量计)。Kizlink[9]采用(Bu2SnO)n做催化剂催化CO2和甲醇反应合成DMC,在反应的过程中(Bu2SnO)n会转化为{Bu2(MeO)Sn-O-Sn(OMe)Bu2}2,并且作者还考察了反应的压力,时间,温度等因素对催化剂活性的影响。根据表征结果和催化剂活性评价结果,其推测了反应机理如图1所示。
该催化过程的主要步骤就是CO2插入到烷氧基和Sn中形成ROC(O)O-Sn,根据催化剂表征,这种单碳酸酯的形成过程比较容易。Choi等[10]用Me2Sn(OMe)2作催化剂,CO2插入到O-Sn,形成结构如图2所示的中间物质,并且该结构已得到了单晶XRD确认。
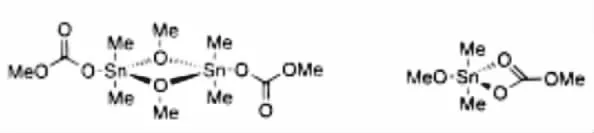
图2 CO2插入到O-Sn形成的中间体
但是有机锡催化剂能够与反应产生的水作用,导致快速失活,同时有机锡催化剂与DMC等产品能形成均相混合物,对催化剂的循环利用和后续产品的分离非常不利。
2.1.2 碱金属催化剂
与有机金属催化剂相比,碱金属催化剂更廉价,毒性也更小。Liu等[11]使用Na2CO3为催化剂催化CO2,甲醇和 PO 合成 DMC,在 0.5MPa,120℃,6h 反应条件下,DMC的收率达到了40%左右。在使用碱金属催化剂的时候,经常会在反应体系中加入CH3I作为促进剂。曹发海[12]选用K2CO3为催化剂的同时在反应体系中加入了CH3I,在高压反应釜中研究了反应温度(60~120℃)和反应压力(4.5~8.0MPa)对液相产品中DMC含量的影响,在373K,7.5MPa的条件下,目标产物DMC在液相产品的质量分数达到的最大值为2.01%。林春绵等[5]在K2CO3和KI催化剂体系的基础上引入了金属醋酸盐,制备了金属醋酸盐与K2CO3、KI复合催化剂,考察了CH3COONa,Ni(CH3COO)2、Zn(CH3COO)2和 CH3COOK 四种醋酸盐的加入对催化剂活性的影响,经过研究发现加入醋酸锌的活性最好,当醋酸锌与K2CO3和KI的质量比为2:1:1时,在合适的反应条件下,PO转化率可达95%,DMC产率可达54.3%。
2.1.3 醋酸盐金属催化剂
醋酸盐属于有机金属化合物,在催化CO2和甲醇直接合成碳酸二甲酯的反应中,表现出了较高的选择性,但催化剂的活性较低。Zhao等[13]人在CO2处于超临界的条件下,研究了CH3COONa,Mn(CH3COO)2,Ni(CH3COO)2、Zn(CH3COO)2和 Co(CH3COO)2等多种醋酸盐的催化活性,根据活性评价结果发现Ni(CH3COO)2催化剂的活性优于其它醋酸盐的催化活性,催化剂的活性达到了每摩尔催化剂796%摩尔DMC。作者同时结合XRD,EXAFS,FT-IR等表征手段提出了Ni(CH3COO)2催化剂的催化机理,如图3。Ni(CH3COO)2催化剂的催化反应过程为:甲醇分子在催化剂表面的碱性位活化为甲氧基,然后催化剂中的Ni与甲氧基结合形成 (NiOCH3)δ+中间体,然后CO2分子插入到Ni-O键中,在经过一系列的表面反应后生成DMC。

图3 有机锡的催化反应机理假设
2.1.4 离子液体催化剂
离子液体是由完全由离子构成的液态熔融盐。1914年,由Walden无意间第一次合成了硝酸乙基铵,这是第一次合成离子液体。离子液体结构和性质独特,能与多种有机物互溶,并且具有良好的导电性,经常用来做电解液。随着越来越多的新型离子液体被合成出来,将其应用于合适的催化反应中也越来越受到研究人员的重视。
Liu等[14]以离子液体1-丁基-3-甲基咪唑四氟硼酸盐为电解质,以In为电极材料在温和实验条件下,考察了其对CO2和甲醇直接合成DMC的电催化性能,当反应温度为40℃,电解电位为-1.7V,电解电量为1.0F/mol时,DMC的产率为76%。Feng等[15]以离子液体BmimBF4为催化剂同时作为电解液,不再添加任何支持电解质,电催化CO2与甲醇合成DMC,在反应温度为55℃时,在5mL离子液体BmimBF4中溶入饱和的CO2,以NPC-Pt为阴极材料,Mg为阳极,Ag丝作为参比电极,电解电位为-2.0V,电解电量为1.0F/mol,甲醇浓度为0.14mol/L,加入3倍甲醇物质的量的碘甲烷,回流反应5h,对分离得到的产物进行分析,最终DMC产率为77.3%。
2.2 多相催化剂
相比于均相催化剂难分离、循环使用次数差等缺点,研究多相催化剂越来越多的成为研究人员关注的热点。
2.2.1 负载型金属有机化合物催化剂
金属有机化合物催化剂虽然在催化CO2和甲醇合成DMC的反应中,具有较高的催化活性,但是其与产物的分离比较困难限制了其使用,为解决这个缺点有研究人员提出采用将金属有机化合物负载到载体上,这样既利于催化剂与产品分离,而且增大了催化剂的循环使用次数。Fan等[16]采用了原位合成的方法制备了有机锡-SBA-15催化剂。有机锡的含量对催化剂的活性有较大的影响,当w(Sn)为1.4%时,在反应条件为:催化剂0.1g,甲醇0.49mol,CO2压 力 18MPa,200℃ ,10h, 催 化 剂 的TON为3.0,相比于四配位的有机锡簇化合物,具有六配位的有机锡簇化合物具有较高的活性。钟等[3]人将Cu2(μ-iOPr)2双核配合物催化剂负载于SiO2载体上,在适当的反应条件下,甲醇的转化率达到5%,而碳酸二甲酯的选择性几乎为100%。作者结合红外、质谱等表征手段,对催化剂的催化机理做出推测,如图4。
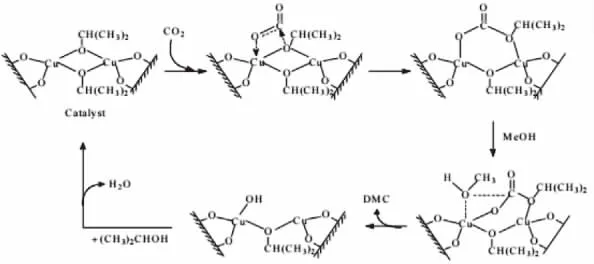
图4 催化剂Cu2(μ-iOPr)2的反应机理
首先CO2在催化剂的表面吸附,主要以桥式吸附态为主,随后发生空间异构变化,转变为异丙氧碳酸酯基吸附结构,而CH3OH在催化剂的表面以分子形式吸附,然后吸附的CH3OH分子亲核进攻异丙氧碳酸酯基吸附态,生成碳酸二甲酯表面物种,最后脱附形成目标产物DMC,生成的表面羟基物种烷氧基化,最终催化剂复原。
2.2.2 负载金属催化剂
Bian等[17]人利用浸渍法制备了活性炭负载的铜基催化剂,对催化剂的结构和性能进行了详细的表征和测试,结果表明在催化剂表面同时存在三种价态的 Cu(Cu0,Cu+,Cu2+),均为催化活性中心,同时催化剂的焙烧温度活极大的影响催化剂表面不同价态的Cu含量,作者研究发现当催化剂的焙烧温度为673K时,催化剂表面的Cu+和Cu2+含量最大,这也促进了催化剂的活性增强。催化剂的活性受Cu的负载量、反应温度等因素影响较大。当负载Cu质量分数为7%,反应温度为393K,DMC的产率最大为 4.77mmol·h-1,当温度继续升高时,副产物(二甲醚、甲酸甲酯)的含量会增加,催化剂选择性下降。
Zhou等[18]使用多种合成方法结合制备了以Mo为助剂的Cu-Fe/SiO2的双金属催化剂。作者首先将(NH4)6Mo7O24浸渍在SiO2载体上,经过焙烧后形成催化剂的前躯体,然后用柠檬酸法将Cu-Fe催化剂负载在 Mo-SiO2催化剂上(w(CuO+Fe2O3)=15%,nCu:nFe=3:2),最后将焙烧过的催化剂用氢气还原。经过对催化剂结构表征发现,助剂Mo能降低Cu-Fe催化剂的还原性,并且增加了Cu-Fe的分散度。通过改变助剂Mo的加入量可以调变催化剂表面的酸碱性,结合CO2,NH3-TPD表征和催化剂活性评价,作者认为中等强度的酸碱性对催化剂的活性是有利的。当w(Mo)为2.5%时,甲醇的转化率达到最大值,为6.99%,DMC的选择性为87.7%。
2.2.3 杂多酸催化剂
Jiang等[19]人用溶胶凝胶法制备了ZrO2负载的具有Keggin结构的磷钨酸(H3PW12O40/ZrO2)。作者考察了反应压力、催化剂的用量、反应温度对DMC产率的影响,发现反应压力对DMC的产率有非常大的影响。当CO2的压力为4 Pa或者7MPa时,DMC的产率最大,可以收集到3mmol的DMC,并且催化剂的选择性非常高,在反应的过程中几乎没有任何的副产物(二甲醚、CO),催化剂用量在0~50mg范围内时,DMC的产率与催化剂的用量有着非常好的线性关系,并且低温有利于DMC的生成。作者结合文献和研究结果提出了相应的催化机理 (图5),研究人员认为催化剂表面的酸碱双性位点是CO2和甲醇直接合成DMC所必需的催化要素,但是H3PW12O40/ZrO2即使在较低的反应温度下仍然极大的促进了DMC的生成,这主要是因为H3PW12O40/ZrO2催化剂的结构,无论是做为载体的ZrO2还是催化剂H3PW12O40/ZrO2,表面都含有Lewis酸位,但是H3PW12O40/ZrO2催化剂表面还包含有较弱的布朗斯特酸性位,相比于路易斯酸位,布朗斯特酸位更有利于甲醇的分子的活化。

图5 H3PW12O40/ZrO2催化CO2和甲醇的可能反应机理
La等[20]利用溶胶凝胶法制备了H3PW12O40/CexTi1-xO2催化剂,并用NH3-TPD,CO2-TPD对催化剂的表面的酸碱性进行了详细的表征和分析。经过活性测试对比在相同的反应条件下,H3PW12O40/CexTi1-xO2催化剂的活性要远远优于CexTi1-xO2催化剂的活性。随着Ce的含量变化,催化剂H3PW12O40/CexTi1-xO2的活性呈现火山型变化。催化剂表面的酸碱性与催化剂的活性有非常强的线性关联,H3PW12O40/CexTi1-xO2催化剂的活性随着催化剂表面的酸碱性的增强而增强。 当 nCe:nTi为 1:9 时,H3PW12O40/Ce0.1Ti0.9O2催化剂的表面具有最强的酸碱性,对应的CO2的吸附量达到最高,DMC的产率最大为5.0mmol。
Lin等[21]人也是使用溶胶凝胶法合成了Ce0.1Ti0.9O2,H3PW12O40/Ce0.1Ti0.9O2催化剂。 研究结果表面,H3PW12O40的加入明显改善了催化剂表面的酸性强度,使催化剂的表面的酸性位的数量增加。在反应温度为 170℃,V(CO2):V(N2)为 1:7 时,催化剂H3PW12O40/Ce0.1Ti0.9O2催化甲醇的转化率达到最大值为5.5%,选择性为91.4%,DMC的产率为为5.0%,均优于催化剂没改性之前的催化剂Ce0.1Ti0.9O2(4.2%,55%,2.3%)。作者运用一阶模型的线性回归方程分别对两个催化剂的速率常数进行了估算,两个催化剂的速率常数分别为:3.34×10-3min-1(Ce0.1Ti0.9O2),4.16×10-3min-1(H3PW12O40/Ce0.1Ti0.9O2),这与催化剂的活性和NH3-TPD的表征结果相对应。作者根据研究结果,结合文献报道对催化剂的催化机理做出了推测和假设,作者认为催化剂表面存在的氧空位在一定程度上能促进甲醇分子和CO2的吸附和活化,并且在生成DMC后,催化剂表面的氧空位能够再生。
2.2.4 金属氧化物催化剂
目前,用于直接催化CO2和甲醇反应的金属氧化物催 化剂主 要包括 :V2O5,MgO,CaO,ZrO2,CeO2等。Meng等[22]人用质量分数为85%磷酸对氧化物V2O5进行改性,发现用磷酸改性过的V2O5催化剂的活性得到了提升。改性催化剂n(P)/n(V)>0.15时,其晶相结构从从单一正交相转变为正交和四方相的混合相,混合相的存在有利与催化剂的活性增强,催化剂表面的弱酸性也得到增强。V和P有利于增强催化剂表面的布朗斯特弱酸性位的生成,V2O5催化剂表面的布朗斯特酸比路易斯酸更有利于甲醇分子的活化。Greish等[23]人在催化剂SnO2/Al2O3里加入Cu,Zn等助剂,助剂的加入有利于催化剂的活性增强,碳酸二甲酯的收率为17.6%,选择性达到了99%。ZrO2,CeO2催化剂表面同时具有路易斯酸碱位,是较早用于催化CO2和甲醇直接合成碳酸二甲酯反应的催化剂。Kaoru Fujimoto等[24]通过直接焙烧ZrO2·xH2O得到ZrO2,研究了焙烧温度对催化剂结构及催化性能的影响。研究结果表明,焙烧温度对催化剂的结构影响非常明显,当焙烧温度为623K时,ZrO2呈现无定型状态,当焙烧温度在673~773K范围内时,催化剂构型呈现亚稳四方相,焙烧温度继续升高催化剂构型呈现单斜相。结合催化剂的活性评价结果,当ZrO2的催化剂构型为亚稳四方相时,催化剂活性高于构型为单斜相的催化剂。Yoshida等[25]同样采用焙烧的方法,在不同的温度下焙烧氢氧化铈得到CeO2催化剂,经过表征和活性评价发现焙烧温度对CeO2的晶型结构和活性同样有着重要的影响。在低于673K的焙烧温度时,CeO2的晶型结构呈现无定型态,没有催化活性,当焙烧温度为873K时,CeO2催化剂呈现立方萤石结构,具有较高的催化活性,在反应温度为423K,催化剂用量为0.1g,反应时间2h时,DMC的产率为0.67mmol。作者同样采用焙烧的方法在ZrO2催化剂中掺杂CeO2,以调变ZrO2催化剂表面的酸碱性。掺杂Ce的ZrO2催化剂催化活性的得到提升,当nCe/n(Ce+Zr)=0.2时,活性最高,DMC的产率为1.6mol。Basudeb Saha等[26]用连续水热回流的方法将CeO2-ZrO2复合氧化物催化剂负载在石墨烯(GO)载体上,制备了具有高活性,高稳定性的催化剂,n(Ce):n(Zr)为1:1。作者使用原甲酸三甲酯作为脱水剂,CeO2-ZrO2/(GO)经过活性测试,甲醇的转化率为58%,DMC的产率为33%,并且经过五次循环后,催化剂的活性并没有明显的下降,说明催化剂具有较好的稳定性。催化剂因形貌不同会暴露不同的晶面,此前有很多文献报道不同形貌的CeO2在不同反应中表现了不同的催化活性。Wang等[27]人利用水热合成法合成了不同形貌的CeO2催化剂:纳米棒,纳米立方体,纳米八面体,纳米纺锤体。经过活性表征发现,不同形貌的CeO2催化剂中,纳米纺锤体的活性最高,其次是纳米棒催化剂,八面体的催化活性最差。作者对催化剂结构研究后认为决定催化剂活性的主要因素是不同形貌的催化剂暴露的晶面不同,导致催化剂表面的酸碱性变化,进而影响了催化剂的活性。纳米纺锤体形貌的CeO2催化剂表面主要暴露的是晶面(111),因此作者认为不同形貌的CeO2催化剂中的活性晶面为(111)面,并结合其它表征结果对CeO2的催化机理提出假设,如图6。

图6 不同形貌CeO2催化CO2和CH3OH的可能的反应机理
3 CO2和甲醇直接合成碳酸二甲酯的反应机理
3.1 路易斯酸碱位机理
根据此前的文献报道,催化剂表面的路易斯酸碱位在催化CO2和甲醇合成DMC的反应中起到非常重要的作用。Alexis T.Bel等[28]人采用原位红外的方法对单斜相ZrO2催化CO2合成DMC的反应机理进行了研究。研究发现甲醇的解离吸附速度慢于CO2的吸附解离速度,但是甲醇吸附解离后的物种在催化剂表面的吸附强度要强于CO2吸附解离物种的强度。甲醇在ZrO2表面的吸附物种为甲氧基,CO2能够插入到甲氧基中形成单甲基碳酸酯中间物种,然后单甲基碳酸酯与另一分子的甲醇反应生成DMC,作者认为CO2分子中的C和O原子以及ZrO2催化剂表面的羟基及催化剂表面配位不饱和的Zr4+和O2-作为路易斯酸碱位,在反应的过程中能够相互产生作用,对催化CO2和甲醇生成DMC起到了关键的作用。Kaoru Fujimoto等[24]人也采用原位红外技术研究了CO2和甲醇在ZrO2催化剂表面的吸附物种,作者认为催化剂表面相邻的酸碱位对DMC的生成有重要的作用。结合文献报道,我们将基于氧化物表面的路易斯酸碱对的催化机理总结在如图7中。
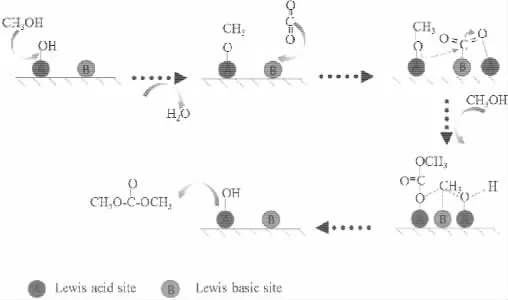
图7 基于催化剂表面路易斯酸碱位催化CO2和甲醇直接合成DMC的反应机理
从图7的机理中,我们看到甲醇分子可以与催化剂表面的羟基作用,在催化剂表面生成吸附的甲氧基的同时脱去了一分子水。CO2分子通过路易斯酸碱对的作用,分子中的碳原子吸附在催化剂表面的碱性位。CO2分子中的碳原子上的电子会向分子中氧原子上转移,同时吸附在相邻酸性位上的甲氧基中的氧原子再次通过路易斯酸碱的作用,使CO2分子插入到甲氧基中氧原子与催化剂表面的路易斯酸位之间,形成了甲基碳酸酯。随后另一分子的甲醇,通过羟基氧中的氧原子与催化剂的表面的路易斯酸位作用,此时甲醇甲基中的碳原子与吸附在路易斯酸位上的甲基碳酸酯的氧原子作用,甲基碳原子上的电子转移至羟基氧上,而甲基碳酸酯吸附在路易斯酸位上的氧原子将电子向甲醇的碳原子转化。经过一系列的表面催化反应后,形成目标产物碳酸二甲酯,并从催化剂表面脱附。
3.2 表面氧空位机理

图8 催化剂表面氧空位催化合成DMC的机理
也有一些研究人员,提出了催化剂表面氧空位在催化CO2和甲醇合成碳酸二甲酯的过程中也起到了重要的作用,这与催化剂表面的路易斯酸碱催化稍有不同(图8)。例如,在催化剂Ce0.1Ti0.9O2或者H3PW12O40改性后Ce0.1Ti0.9O2表面氧空位的反应机理。有文献报道[19],催化剂表面的氧空位相当于路易斯酸位,可以与CO2分子中的氧原子产生作用。从图8我们看到,CO2分子通过氧原子插入到催化剂表面的氧空位中,吸附在催化剂的表面。另一分子的甲醇羟基中的氢原子插入到相邻的氧空位中,随后另一分子甲醇中羟基氧的氢原子插入到另一相邻的表面氧空位中,CO2和甲醇的吸附会在催化剂表面形成一系列不稳定的中间吸附物种,然后经过一系列的催化剂表面的催化反应,这些吸附的中间产物转化为碳酸二甲酯和水,并从催化剂Ce0.1Ti0.9O2或者H3PW12O40/Ce0.1Ti0.9O2表面脱附,表面氧空位再生,进而完成一个催化反应的循环。在整个催化反应进行的过程中作者也观察到,表面氧空位随着反应的进行会部分被消耗掉,进而导致催化活性下降。
4 结语
二氧化碳作为引起温室效应的主要气体,同时又是一种非常廉价和丰富的碳源。现在世界各国都非常重视绿色化学、经济化学的利用,有效捕集和利用CO2已越来越引起科研工作者的关注。将CO2与廉价易得的化工产品甲醇直接合成DMC反应,因其原子利用率高、合成路线绿色环保,近年来成为研究的的热点。由于热力学上的限制CO2分子的惰性,通过CO2和甲醇直接合成碳酸二甲酯反应的平衡转化率和平衡常数都非常小。因此除了研发高效的催化剂外,研究人员应试图研究新型的反应体系,对反应物或者产物进行一定的转化,以突破热力学对该反应的限制,促使反应向右进行,增加碳酸二甲酯的产率。
参考文献
[1] Artz J,Müller T E,Th enert K,et al.Sustainable conversion of carbon dioxide:An integrated review of catalysis and life cycle assessment[J].Chem Rev,2018,118:434-504.
[2] Ono Y.Catalysis in the production and reactions of dimethyl carbonate,an environmentally benign building block[J].Appl Catal A,1997,155(2):133-166.
[3] 孔令丽,钟顺和,肖秀芬.超临界条件下负载型配合物催化剂Cu2(μ-OEt)2/SiO2合成碳酸二甲酯的反应性能[J].分子催化,2004,18(3):172-178.
[4] 孔令丽,钟顺和.负载型双核配合物 Cu2(μ-iOPr)2/SiO2的制备与表征及超临界条件下合成DMC的反应性能[J].高校化学工程学报,2008,18(6):701-706.
[5] 林春绵,丁春晓,张平等.金属醋酸盐复配催化剂催化超临界CO2一步法合成碳酸二甲酯[J].高校化学工程学报,2012,26(2):320-325.
[6] Ding L,Fan X,Sun X M,et al.Direct preparation of semiconductor iron sulfide nanocrystals from natural pyrite[J].RSCAdv,2013,3:4539-4543.
[7] Li C F,Zhong S H.Study on application of membrane reactor in direct synthesis DMC from CO2and CH3OH over Cu-KF/MgSiO catalyst[J].Catal Today,2003,82:83-90.
[8] Kizlink J,Pastucha I.Preparation of dimethyl carbonate from methanol and carbon dioxide in the presence of Sn(IV)and Ti(IV)alkoxides and metal acetates[J].Collect Czechoslovak Chem Commun,1994,59(9):2116-2118.
[9] Kizlink J.Synthesis of dimethyl carbonate from carbon dioxide and methanol in the presence of organotin compounds[J].Collect Czech Chem Commun,1993,58:1399-1402.
[10]Choi J C,Kohno K,Ohshima Y,et al.Tin-or titaniumcatalyzed dimethyl carbonate synthesis from carbon dioxide and methanol:Large promotion by a small amount of triflate salts[J].Catal Commun,2008,9:1630-1633.
[11]Liu C,Zhang S K,Cai B Y,et al.Low pressure one-pot synthesis of dimethyl carbonate catalyzed by an alkali carbonate[J].Chin JCatal,2015,36(7):1136-1141.
[12]曹发海,刘殿华,房鼎业.碱性催化剂作用下CO2与甲醇直接合成碳酸二甲酯的探索性研究 [J].化学世界,2000,41(11):594-596.
[13]Zhao T S,Han Y Z,Sun Y H.Novel reaction route for dimethyl carbonate synthesis from CO2and methanol[J].Fuel Process Technol,2000,62:187-194.
[14]Feng Q J,Huang K L,Liu S Q,et al.Electrocatalytic carboxylation of aromatic ketones with carbon dioxide in ionic liquid 1-butyl-3-methylimidazoliumtetrafluoborate to α-hydroxy-carboxylic acid methyl ester[J].Electrochimica Acta,2011,5:5741-5745.
[15]Feng Q J,Huang K L,Liu S Q,et al.Electrocatalytic carboxylation of 2-amino-5-bromopyridine with CO2in ionic liquid 1-butyl-3-methyllimidazoliumtetrafluoborate to 6-aminonicotinic acid[J].Electrochimica Acta,2010,55:5741-5745.
[16]Fan B,Zhang J,Li R,et al.In situ preparation of functional heterogeneous organotin catalyst tethered on SBA-15[J].Catal Lett,2007,121:297-302.
[17]Bian J,Wei X W,Jin Y R,et al.Direct synthesis of dimethyl carbonate over activated carbon supported Cubased catalysts[J].Chem Eng J,2010,165:686-692.
[18]Zhou Y J,Xiao M,Wang S J,et al.Effects of Mo promoters on the Cu-Fe bimetal catalysts for the DMC formation from CO2and methanol[J].Chin Chem Lett,2013,24(4):307-310.
[19]Jiang C,Guo Y,Wang C,et al.Synthesis of dimethyl carbonate from methanol and carbon dioxide in the presence of polyoxometalates under mild conditions[J].Appl Catal A,2003,256:203-212.
[20]La K W,Ji C J,Kim H,et al.Effect of acid-base properties of H3PW12O40/CexTi1-xO2catalysts on the direct synthesis of dimethyl carbonate from methanol and carbon dioxide:A TPD study of H3PW12O40/CexTi1-xO2catalysts[J].JMol Catal A,2007,269:41-45.
[21]Chiang C L,Lin K S,Yu S H,et al.Synthesis and characterization of H3PW12O40/CexTi1-xO2for dimethyl carbonate formation via Methanol carbonation[J].Int J Hydrogen Energy,2017,42:22108-22122.
[22]Wu X L,Xiao M,Meng Y Z,et al.Direct synthesis of dimethyl carbonate on H3PO4modified V2O5[J].JMol Catal A,2005,238:158-162.
[23]Greish A A,Finashina E D,Tkachenko O P,et al.Synthesis of dimethyl carbonate from methanol and CO2on the SnO2/Al2O3-based catalyst[J].Mendeleev Commun,2016,26:497-499.
[24]Tomishige K,Sakaihori T,Ikeda Y,et al.A novel method of direct synthesis of dimethyl carbonate from methanol and carbon dioxide catalyzed by zirconia[J].Catal Lett,1999,58:225-229.
[25]Yoshida Y,Arai Y,Kado S,et al.Direct synthesis of organic carbonates from the reaction of CO2with methanol and ethanol over CeO2catalysts[J].Catal Today,2006,115:95-101.
[26]Saada R,Kellici S,Heil T,et al.Greener synthesis of dimethyl carbonate using a novel ceria-zirconia oxide/graphene nanocomposite catalyst[J].Appl Catal B,2015,168-169:353-362.
[27]Wang S,Zhao L,Wang W,et al.Morphology control of ceria nanocrystals for catalytic conversion of CO2with methanol[J].Nanoscale,2013,5:5582-5588.
[28]Jung K T,Bell A T.An in situ infrared study of dimethyl carbonate synthesis from carbon dioxide and methanol over zirconia[J].JCatal,2001,204:339-347.
猜你喜欢
科学大众(2023年17期)2023-10-26 07:38:56
云南化工(2021年10期)2021-12-21 07:33:42
小天使·二年级语数英综合(2021年5期)2021-07-11 10:58:35
云南化工(2020年11期)2021-01-14 00:50:48
应用化工(2020年9期)2020-09-29 08:55:16
中学化学(2017年2期)2017-04-01 08:51:54
河南科技(2015年2期)2015-02-27 14:20:35
中南民族大学学报(自然科学版)(2014年4期)2014-08-06 05:49:24
同位素(2014年2期)2014-04-16 04:57:13
无机化学学报(2014年7期)2014-02-28 17:32:10
