
盐酸羟胺强化Fe(Ⅲ)-EDDS/过硫酸盐处理水溶液中TCE
2018-04-25 13:08顾小钢上海市城市建设设计研究总院集团有限公司土壤修复研究中心上海200125
中国环境科学 2018年4期
顾小钢 (上海市城市建设设计研究总院(集团)有限公司,土壤修复研究中心,上海 200125)
近年来,随着我国城市化和工业化快速发展,城市中遗留了大量污染场地.氯代烃(如三氯乙烯,TCE)作为重要的化工原料和有机溶剂,由于生产、使用、存储等过程的不当操作[1],导致其成为污染场地检出率较高的有机污染物之一.
过硫酸盐(PS)是一种新型氧化剂,广泛用于治理场地有机污染物.过硫酸盐常温下比较稳定,在热、光、过渡金属、碱、过氧化氢等活化条件下,能够产生硫酸根自由基(SO4•−)、羟基自由基(•OH)、超氧阴离子自由基(O2•−)等活性氧自由基(ROS),从而有效降解目标污染物[2-7].与芬顿反应相似,Fe(Ⅱ)活化过硫酸盐(简写为Fe(Ⅱ)/PS,式1)具有速度快、成本低、易操作等优点,能够有效去除水溶液中的 TCE.但是,Fe(Ⅱ)直接活化过硫酸盐过程中铁有效性较低,存在 pH值适用范围窄、活性自由基易被过量Fe(Ⅱ)消耗、持续活化能力差等不足[8-9].
研究表明,加入螯合剂能可以提高铁有效性,强化过硫酸盐活化效率[10].乙二胺四乙酸(EDTA)是最常用的金属螯合剂之一,但其自身具有毒性且难以生物降解.S,S'-乙二胺-N,N'-二琥珀酸三钠(EDDS)是EDTA的同分异构体,具有易生物降解、环境友好的特点[11].作者前期研究发现,EDDS螯合Fe(Ⅲ)能够显著提高Fe(Ⅲ)/PS降解水溶液中 TCE的效率[12].但是,Fe(Ⅲ)-EDDS/PS体系受溶液 pH 值、Cl−和 HCO3−的影响仍比较明显.
铁活化高级氧化体系中加入还原性物质,如盐酸羟胺、硫代硫酸钠、抗坏血酸等,能够促进活化体系中 Fe(Ⅱ)循环再生,从而改善氧化剂活化效率及污染物去除效率[13].盐酸羟胺自身与氧化性自由基反应速率较低,且反应后生成的最终产物为无机物质,常用于强化铁活化高级氧化体系[12].研究表明,加入HAH能够显著提高Fenton或铁活化过碳酸盐氧化效率[13,15].此外,HAH也能够促进 Fe(Ⅱ)活化过硫酸盐工艺降解有机污染物[14,16-17].目前,尚无 HAH 强化 Fe(Ⅲ)活化过硫酸盐的研究.因此,本研究拟在前期关于Fe(Ⅲ)-EDDS/PS去除TCE的基础上,考察HAH强化Fe(Ⅲ)-EDDS/PS工艺降解TCE的效果,分析工艺参数(PS和 HAH初始浓度)及水质条件(初始pH值和阴离子)的影响,最后验证强化体系中活性氧自由基类型(主要考察 SO4•−、•OH 和O2•−),并探究其对TCE 降解的贡献度.
1 材料与方法
1.1 试剂及仪器
试剂:过硫酸钠、三氯乙烯、四氯化碳(CT)均为分析纯,购自国药集团化学试剂有限公司.EDDS(35%水溶液)购自 Sigma-Aldrich上海公司,盐酸羟胺、硫酸铁、正己烷、硝基苯、苯甲醚、异丙醇、叔丁醇、1,4-苯醌、氯化钠、碳酸氢钠、硫酸、氢氧化钠均为分析纯,购自阿拉丁试剂(试剂)有限公司.
主要仪器:气相色谱仪(Agilent 7890A)、磁力搅拌器(85-2,上海闵行虹浦仪器厂)、低温恒温槽(SDC-6,宁波新芝生物科技股份有限公司)、pH计(Mettler-Toledo DELTA320)、超纯水仪(ELGA Classic DI).
1.2 试验方法
采用带夹层的玻璃制圆柱体(内径 5cm、高12cm、有效容积250mL)作为反应器.反应器置于磁力搅拌器上,转速恒定为 600r/min,采用低温恒温槽控制反应温度为(20±0.5)℃.硫酸铁与EDDS混合搅拌15min后,将其加入TCE水溶液,最后投加过硫酸盐开始反应,反应液总体积为250mL(实验时间内 TCE挥发量<3%).既定时间取样分析,每组实验设 2个平行.考察溶液初始pH值影响时,采用 0.1mol/L NaOH和 0.1mol/L H2SO4调节,其他反应均不调节溶液 pH 值.所有溶液均采用超纯水配制,TCE初始浓度为0.15mmol/L.根据前期研究结果,本研究中Fe(Ⅲ)/EDDS物质的量比设为 4/1,Fe(Ⅲ)初始浓度为1.8mmol/L.
1.3 分析方法
试验中采用气相色谱仪测定三氯乙烯、四氯化碳、硝基苯、苯甲醚浓度.三氯乙烯和四氯化碳采用DB-VRX色谱柱(长60m、内径250µm、膜厚1.4µm)、ECD检测器分析,进样口与检测器温度分别为 240、260℃,柱温 75℃,进样量 1µL,分流比 20:1.硝基苯和苯甲醚采用 HP-5色谱柱(长30m、内径250µm、膜厚0.25µm)、FID检测器分析,进样口与检测器温度分别为200、250℃,柱温150℃,进样量1µL,分流比20:1.过硫酸盐和溶解性铁离子浓度分别采用碘化钾、邻菲啰啉分光光度法测定.
2 结果与讨论
2.1 HAH强化Fe(Ⅲ)-EDDS/PS降解TCE的效果
图 1显示了 TCE 在 Fe(Ⅲ)/PS、Fe(Ⅲ)-EDDS/PS和Fe(Ⅲ)-EDDS/PS/HAH体系中的降解效果, 过硫酸盐和盐酸羟胺投加量均为7.5mmol/L.Fe(Ⅲ)/PS体系中,60min内TCE去除率为 12.8%,表明 Fe(Ⅲ)直接活化过硫酸盐效率较低.Fe(Ⅲ)-EDDS/PS体系中,反应前0.5min内TCE去除率为12.4%,之后TCE去除率缓慢升高至75.3%(60min),表明加入EDDS能够有效提高溶液中铁有效性,提高 TCE去除率.Fe(Ⅲ)-EDDS/PS体系中进一步加入 HAH后,能够明显强化TCE去除效率.60min内TCE基本完全降解,且前 0.5min内去除率达 89.0%.由此推断,加入HAH能够促进Fe(Ⅲ)还原为Fe(Ⅱ),进而强化过硫酸盐活化效果.Zou等[18]研究发现HAH不仅能加速 Fe(Ⅱ)/Fe(Ⅲ)循环,还可以强化活性氧自由基产率,从而促进 Fe(Ⅱ)活化过一硫酸盐降解苯甲酸.

图1 HAH强化Fe(Ⅲ)-EDDS/PS降解TCE的效果Fig.1 HAH enhancing Fe(Ⅲ)-EDDS/PS for TCE degradation
2.2 过硫酸盐、盐酸羟胺初始浓度的影响
Fe(Ⅲ)/EDDS物质的量比设为4/1,考察过硫酸盐和盐酸羟胺初始浓度对TCE降解效果的影响.由图2a可以看出,不投加过硫酸盐时TCE基本不发生降解,过硫酸盐浓度由1.50mmol/L增加到7.50mmol/L时,TCE去除率随过硫酸盐浓度升高而增强,60min内去除率分别达71.2%、93.8%和99.7%.继续增加过硫酸盐浓度至11.25mmol/L,TCE降解效果没有进一步增强.因此后续实验将过硫酸盐初始浓度设为7.50mmol/L.
HAH除了能够促进Fe(Ⅱ)/Fe(Ⅲ)循环,还能通过非自由基途径消耗过硫酸盐,并且 HAH中的 Cl−可能与 SO4•−反应生成低活性自由基[16].因此,考察了HAH初始浓度对TCE降解效果的影响,结果如图2b所示.实验条件下,加入HAH显著提高了 Fe(Ⅲ)-EDDS/PS工艺降解 TCE的效率,TCE去除率随 HAH浓度(1.50~7.50mmol/L)增大而升高.HAH初始浓度为 7.50mmol/L时,60min内TCE基本完全去除.继续增加HAH投加量至11.25mmol/L,0~10min内TCE降解效率受到轻微抑制,10~60min 降解效果与7.50mmol/L相近.因此,适量的 HAH 能够促进Fe(Ⅱ)再生,有利于活化过硫酸盐,而 HAH 浓度过高时可能抑制 TCE降解,后续实验中将 HAH浓度设为7.50mmol/L.
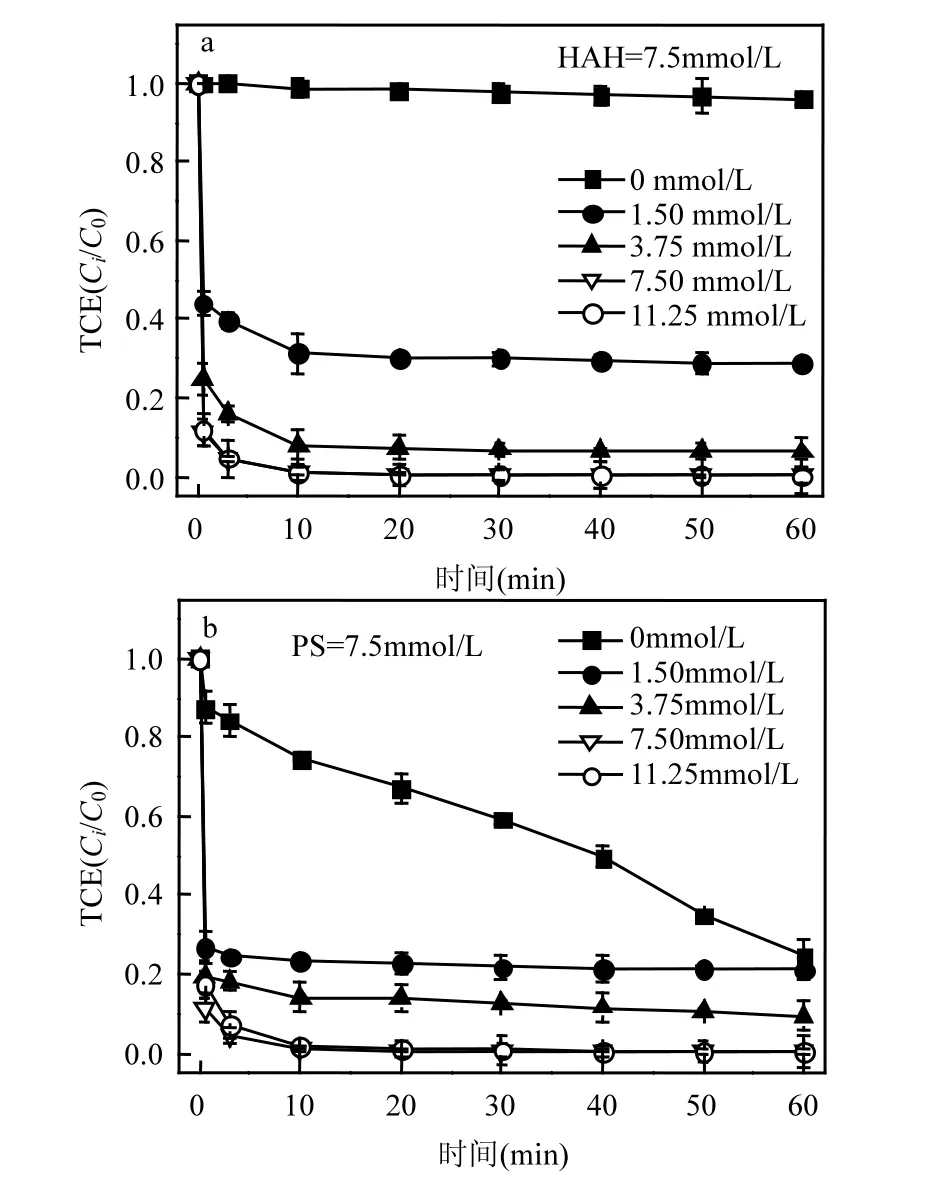
图2 PS、HAH初始浓度对TCE降解效果的影响Fig.2 Effects of PS or HAH initial concentration on TCE degradation
2.3 溶液初始pH值的影响
考察了溶液(含 Fe(Ⅲ)-EDDS、TCE、HAH的溶液)初始pH值对Fe(Ⅲ)-EDDS/PS/HAH降解TCE的影响(图3).原始溶液初始pH值为2.7,加入过硫酸盐反应60min后降至1.6.实验调节初始pH值为3、5、7时,对TCE降解基本没有影响,反应60min后TCE均基本完全去除,此时溶液pH值下降至2.0~2.2.但是,调节溶液初始pH值为8、9、11,反应终止时 TCE去除率均低于 30%,而此时溶液pH值也维持在较高值(6.4~7.2).由此可见,初始溶液为酸性或中性,反应终止时 pH<3,对Fe(Ⅲ)-EDDS/PS/HAH降解TCE基本没有影响;而当溶液初始 pH值为碱性(pH=8、9或 11)时, TCE降解受到明显抑制,且在这种情况下,反应终止时溶液pH值均<6.

图3 溶液初始pH值对TCE降解效果的影响Fig.3 Effect of initial solution pH on TCE degradation
2.4 Cl−和 HCO3−的影响
由于Cl−和HCO3−均能够消耗体系中产生的SO4•−或•OH[19],因此选择 Cl−和 HCO3−为代表,考察地下水阴离子对 Fe(Ⅲ)-EDDS/PS/HAH降解TCE的影响.作者前期研究发现[12], Fe(Ⅲ)-EDDS/PS体系中加入 1~100mmol/L 的 Cl−后,TCE降解率由97.0%下降至30.9%.但在本研究中,加入低浓度 Cl−(1、10mmol/L),对 TCE 降解基本没有影响(图 4a).Cl−投加量为 100mmol/L时,60min内 TCE去除率仍达 92.6%.表明在Fe(Ⅲ)-EDDS/PS体系加入HAH后,不仅能够有效促进TCE降解,同时能够降低Cl−对TCE降解的抑制作用.
图 4b 为 HCO3−对 Fe(Ⅲ)-EDDS/PS/HAH 降解 TCE 的影响.当 HCO3−浓度为 1、10mmol/L时,60min内 TCE降解基本不受影响,此时溶液pH 值均<2.5.增加 HCO3−浓度至 25、50、100mmol/L则明显抑制TCE降解,60min内TCE去除率分别为 27.7%、23.7%、19.8%,并且反应终止时溶液pH值均>6.反应终止时TCE去除率和溶液 pH值的关系与 2.3节结论一致.因此,Fe(Ⅲ)-EDDS/PS/HAH体系中加入高浓度HCO3−(25~100mmol/L)不仅会消耗活性氧自由基,同时能够改变溶液pH值,从而抑制TCE降解.需要注意的是,Fe(Ⅲ)-EDDS/PS体系中加入10mmol/L的HCO3−能够明显抑制TCE降解[12],而加入 HAH后能够缓解低浓度 HCO3−对 TCE降解的抑制作用.
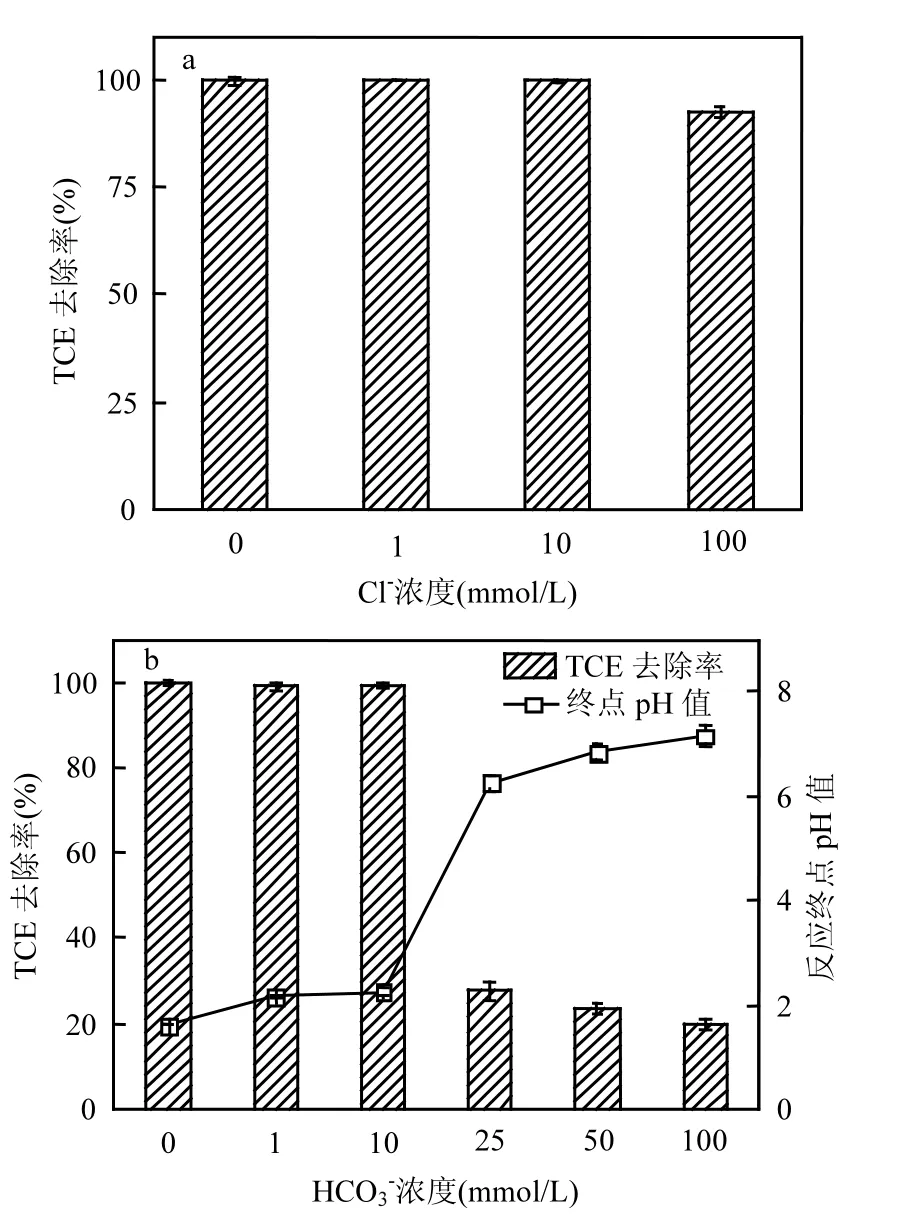
图4 Cl−和HCO3−对TCE降解效果的影响Fig.4 Effect of Cl− or HCO3− on TCE degradation
2.5 活性氧自由基鉴定
2.5.1 化学探针降解实验(无 TCE) 通过Fe(Ⅲ)-EDDS/PS/HAH 体系中自由基探针降解情况,判断可能存在的活性氧自由基.选择硝基苯作 为•OH 探 针 (k•OH=3.9×109L/(mol·s), kSO4•−<106L/(mol·s))[20],苯甲醚作为 SO4•−和•OH 的共同探 针 (k•OH=6.0×109L/(mol·s), kSO4•−=4.9×109L/(mol·s))[20],四 氯 化 碳 作 为 O2•−探 针 (kO2•−=1.6×1010L/(mol·s))[21],硝基苯、苯甲醚、四氯化碳初始浓度分别为2、2、0.05mmol/L.反应时间为240min,结果如图5所示.前期空白实验表明,硝基苯与苯甲醚在反应时间内基本无挥发,四氯化碳空白挥发损失 9.1%.240min内,硝基苯去除率为16.5%,推断体系中存在•OH;苯甲醚去除率为25.9%,则反应过程中至少存在 SO4•−或•OH 中的一种.为进一步验证 SO4•−,在苯甲醚降解过程中加入过量的叔丁醇(2mol/L),以完全消耗溶液中的•OH,而不消耗 SO4•−(k•OH=5.2× 108L/(mol·s),kSO4•−<106L/(mol·s))[20].结果显示,加入叔丁醇后苯甲醚降解效果受到抑制,但仍有17%去除率,表明该体系中同时存在 SO4•−和•OH.反应终止时,四氯化碳去除率为 69.1%(其中挥发损失 9.1%),表明 Fe(Ⅲ)-EDDS/PS/HAH 能够产生 O2•−.Miao等[15]研究了多种还原剂对 Fe(Ⅱ)、Fe(Ⅲ)活化过碳酸钠降解四氯乙烯的影响,发现加入 HAH对生成O2•−具有促进作用.

图5 自由基探针的降解效果Fig.5 The degradation performance of radical probe compounds
2.5.2 自由基清扫实验(含 TCE) 为进一步判断 SO4•−、•OH、O2•−各自对 TCE 降解的贡献情况,在Fe(Ⅲ)-EDDS/PS/HAH降解TCE体系中针对性的加入自由基清扫剂,通过对TCE降解的抑制程度判断各自由基的贡献度.选择叔丁醇作为•OH 清扫剂(k•OH=5.2×108L/(mol·s), kSO4•−<106L/(mol·s)),异丙醇作为 SO4•−和•OH 共同清扫剂(k•OH=2.0×109L/(mol·s),kSO4•−=8.2×107L/(mol·s))[20],1,4- 苯 醌 作 为 O2•−清 扫 剂 (kO2•−=9.6×108L/(mol·s))[22],叔丁醇、异丙醇、1,4-苯醌初始浓度分别为300、500、50mmol/L.反应时间为60min,结果如图 6所示.前期实验表明清扫剂浓度均过量,可认为能够完全清扫相对应的自由基.与控制组相比,加入叔丁醇后TCE去除率下降了28.5%(99.7%~71.2%),由此推断•OH 对 TCE降解的贡献度为 28.5%.加入 IPA后 TCE去除抑制率为78.0%(99.7%~21.7%),表明SO4•−与•OH的总贡献度为 78.0%,则 SO4•−贡献度为 49.5%(78.0%~28.5%).假定 TCE 降解只受 SO4•−、•OH、O2•−作用,则可推断出 O2•−贡献度为 22.0%(100%~78%).但是,加入 1,4-苯醌后 TCE去除率与控制组相比下降了 48.5%(99.7%~51.2%),与异丙醇实验组推算结果存在差异.可能是由于1,4-苯醌消耗了部分 SO4•−或•OH,导致 TCE 去除率下降,这也与作者前期的试验结论一致[12].因此,Fe(Ⅲ)-EDDS/PS/HAH 体系中 TCE 降解由 SO4•−、•OH、O2•−共同作用所致,其中 SO4•−为主导自由基,根据清扫实验推算结果,SO4•−、•OH、O2•−的贡献度分别为49.5%、28.5%和22.0%.需要指出的是,Fe(Ⅲ)-EDDS/PS体系中 TCE降解的主导自由基为•OH[12],加入 HAH 后,主导自由基转变为SO4•−.

图6 自由基清扫剂对TCE降解效果的影响Fig.6 Effect of radical scavengers on TCE degradation
3 结论
3.1 Fe(Ⅲ)-EDDS/PS体系中加入HAH能够强化TCE去除效率.
3.2 溶液初始pH值为酸性或中性时,TCE去除几乎没有影响,碱性条件下TCE降解受到抑制.
3.3 Cl−(1~100mmol/L)和 低 浓 度 HCO3−(1~10mmol/L)对 TCE降解基本没有影响,高浓度HCO3−(25~100mmol/L)抑制效果明显.
3.4 Fe(Ⅲ)-EDDS/PS体系中加入HAH后SO4•−产率增强,TCE降解的主导自由基由•OH转变为SO4•−.
参考文献:
[1]陆 强,李 辉,林匡飞,等.上海浦东某氯代烃场地地下水污染现状调查 [J]. 环境科学学报, 2016,36(5):1730-1737.
[2]Matzek L W, Carter K E. Activated persulfate for organic chemical degradation: A review [J]. Chemosphere, 2016,151:178-188.
[3]朱思瑞,高乃云,鲁 仙,等.热激活过硫酸盐氧化降解水中双酚A [J]. 中国环境科学, 2017,37(1):188-194.
[4]骆靖宇,李学艳,李青松,等.紫外活化过硫酸钠去除水体中的三氯卡班 [J]. 中国环境科学, 2017,37(9):3324-3331.
[5]张丽娜,钟 华,张俊涛,等.热,碱和 Fe3O4激活过硫酸钠降解二恶烷的对比研究 [J]. 中国环境科学, 2017,37(10):3741-3747.
[6]郭佑罗,关小红,高乃云,等.紫外/过硫酸盐工艺降解水中氯贝酸的研究 [J]. 中国环境科学, 2016,36(7):2014-2019.
[7]刘佳露,卢 伟,张凤君,等.活化过硫酸盐氧化地下水中苯酚的动力学研究 [J]. 中国环境科学, 2015,35(9):2677-2681.
[8]Ahn S, Peterson T D, Righter J, et al. Disinfection of ballast water with iron activated persulfate [J]. Environmental Science &Technology, 2013,47(20):11717-11725.
[9]Liang C, Bruell C J, Marley M C, et al. Persulfate oxidation for in situ remediation of TCE. I. Activated by ferrous ion with and without a persulfate–thiosulfate redox couple [J]. Chemosphere,2004,55(9):1213-1223.
[10]Wu X, Gu X, Lu S, et al. Degradation of trichloroethylene in aqueous solution by persulfate activated with citric acid chelated ferrous ion [J]. Chemical Engineering Journal, 2014,255:585-592.
[11]Huang W, Brigante M, Wu F, et al. Assessment of the Fe(III)–EDDS complex in fenton-like processes: From the radical formation to the degradation of bisphenol A [J]. Environmental Science & Technology, 2013,47(4):1952-1959.
[12]Gu X, Wang Y, Miao Z, et al. Degradation of trichloroethylene in aqueous solution by persulfate activated with Fe(III)–EDDS complex[J]. Research on Chemical Intermediates, 2017,43(1):1-13.
[13]Chen L, Ma J, Li X, et al. Strong enhancement on fenton oxidation by addition of hydroxylamine to accelerate the ferric and ferrous iron cycles [J]. Environmental Science & Technology,2011,45(9):3925-3930.
[14]Wu X, Gu X, Lu S, et al. Strong enhancement of trichloroethylene degradation in ferrous ion activated persulfate system by promoting ferric and ferrous ion cycles with hydroxylamine [J].Separation and Purification Technology, 2015,147:186-193.
[15]Miao Z, Gu X, Lu S, et al. Enhancement effects of reducing agents on the degradation of tetrachloroethene in the Fe(II)/Fe(III)catalyzed percarbonate system [J]. Journal of Hazardous Materials, 2015,300:530-537.
[16]Han D, Wan J, Ma Y, et al. Enhanced decolorization of orange G in a Fe(II)-EDDS activated persulfate process by accelerating the regeneration of ferrous iron with hydroxylamine [J]. Chemical Engineering Journal, 2014,256:316-323.
[17]Tan C, Gao N, Chu W, et al. Degradation of diuron by persulfate activated with ferrous ion [J]. Separation and Purification Technology, 2012,95:44-48.
[18]Zou J, Ma J, Chen L, et al. Rapid acceleration of ferrous iron/peroxymonosulfate oxidation of organic pollutants by promoting Fe(III)/Fe(II) cycle with hydroxylamine [J].Environmental Science & Technology, 2013,47(20):11685-11691.
[19]Criquet J, Leitner N K V. Degradation of acetic acid with sulfate radical generated by persulfate ions photolysis [J]. Chemosphere,2009,77(2):194-200.
[20]Liang C, Su H-W. Identification of sulfate and hydroxyl radicals in thermally activated persulfate [J]. Industrial & Engineering Chemistry Research, 2009,48(11):5558-5562.
[21]Lutze H V, Bakkour R, Kerlin N, et al. Formation of bromate in sulfate radical based oxidation: Mechanistic aspects and suppression by dissolved organic matter [J]. Water Research,2014,53:370-377.
[22]Monteagudo J M, Durán A, San Martin I, et al. Roles of different intermediate active species in the mineralization reactions of phenolic pollutants under a UV-A/C photo-fenton process [J].Applied Catalysis B: Environmental, 2011,106(1/2):242-249.
猜你喜欢
中国资源综合利用(2022年9期)2022-10-13
材料与冶金学报(2022年2期)2022-08-10
现代矿业(2022年3期)2022-04-09
建材发展导向(2021年15期)2021-11-05
科学与财富(2021年33期)2021-05-10
作文成功之路·小学版(2020年6期)2020-07-27
作文成功之路·小学版(2020年5期)2020-06-11
制造技术与机床(2019年9期)2019-09-10
同济大学学报(自然科学版)(2019年8期)2019-08-07
中国造纸(2014年1期)2014-03-01
