
液体糖中还原糖含量测定方法的研究*
2018-04-10 06:31陈志颖徐正康叶晓蕾李海燕周彦斌
食品工程 2018年1期
陈志颖 徐正康 郭 峰 叶晓蕾 李海燕 周彦斌
(广州双桥股份有限公司,广东广州 510280)
液体糖以白砂糖、绵白糖、精制的糖蜜或中间制品为原料,经加工或转化工艺制炼而成,可替代白砂糖应用于食品工业,在欧美国家,液体糖的生产和应用已有悠久的历史。
液体糖按糖分组成可分为两大类:全蔗糖糖浆和转化糖浆,其中全蔗糖糖浆的固形物含量≥65.0%,干物质中总糖量≥99.5%;转化糖浆固形物含量≥70.0%,干物质中总糖量≥99.5%,干物质中还原糖≥60.0%。
在QB/T 4093—2010《液体糖》标准中定义了转化糖浆干物质中还原糖含量的2种测定方法,分别为QB/T2343.2—2013《赤砂糖试验方法》中的兰-艾农恒容法和GB/T 18932.22—2003的液相色谱示差折光检测法,但是未见有关全蔗糖糖浆干物质中还原糖含量测定方法的相关描述。本文以此为背景,试验探究转化糖浆干物质中还原糖含量的2种测定方法应用于全蔗糖糖浆干物质中还原糖含量测定的合理性,并提出一种新的检测方法——离子色谱法,分析比对这3种方法应用于全蔗糖糖浆中还原糖含量测定的操作差异性及其检测结果的可靠性。
1 材料与方法
1.1 材料与试剂
材料:液体糖(全蔗糖糖浆),广州双桥股份有限公司生产。
试剂:蔗糖、草酸钾、亚甲基蓝、硫酸铜、酒石酸钾钠、氢氧化钠、无水乙醇、碳酸钠、盐酸、酚酞、苯甲酸、甲基橙,均为分析纯,广州化学试剂厂;果糖、葡萄糖(标准物质),美国Sigma公司。
1.2 仪器与设备
DR-A1阿贝折光仪,日本ATAGO株式会社;S210型pH计,美国梅特勒公司;2414高效液相色谱仪,美国waters公司;Rezex系列钙离子色谱柱(300 mm×7.8 mm),美国Phenomenex公司;AS系列超声波清洗机,天津奥特赛恩仪器有限公司;Millipore超纯水仪,美国密理博公司;930系列离子色谱仪,瑞士Metrohm公司;DionexTMCarboPacTMPA1分析和保护柱(4 nm×250 nm),美国Thermo Fisher Scientific公司;AREC.T加热磁力搅拌器,意大利VELP公司;MCP500高精度数字式旋光仪,奥地利Anton Paar公司。
1.3 试验方案
1.3.1 滴定法用试剂准备
1.3.1.1 10 g/L标准转化糖液
称取蔗糖23.75 g,用蒸馏水约120 mL溶解并移入250 mL的容量瓶中,加入浓盐酸9 mL,摇匀,在室温20℃~25℃下静置8 d,然后用蒸馏水稀释至刻度。吸取该溶液100 mL(含10 g转化糖) 于1 000 mL容量瓶中,在不断摇荡下,加入1 mol/L氢氧化钠溶液,调节pH至3.0(加碱量的确定:另取转化糖液50 mL,以甲基橙作指示剂,以1 mol/L氢氧化钠溶液滴定至红色恰好变为橙色为止,所耗用的氢氧化钠溶液的量乘2即为要加入的碱量),调节pH后加入已用热水溶解的苯甲酸2 g,摇匀,冷却后稀释至刻度。此溶液每100 mL含1 g转化糖,可作为稳定的贮备液。
1.3.1.2 2.5 g/L标准转化糖液
准确吸取10 g/L标准转化糖液50 mL,移入200 mL容量瓶中,加酚酞指示液5滴,在不断摇荡下滴入0.5 mol/L氢氧化钠溶液,直至浅红色出现不褪色为止,加水稀释至刻度,摇匀。
1.3.1.3 费林氏试剂
费林氏甲液:称取硫酸铜69.28 g,用蒸馏水溶解后,移入1 000 mL容量瓶中,加入至刻度,摇匀,过滤即成。
费林氏乙液:称取酒石酸钾钠346 g溶于约500 mL蒸馏水中;另称取氢氧化钠100 g溶于约200 mL蒸馏水中,将二者混合,移入1 000 mL容量瓶中,加水至刻度,放置2 d。如液面降低,应补加至刻度,摇匀,过滤即成。
1.3.1.4 50 g/L草酸钾溶液
称取草酸钾50g加蒸馏水溶解后稀释至1000mL。
1.3.1.5 10 g/L亚甲基蓝溶液
称取亚甲基蓝1.0 g,加蒸馏水溶解后定溶于100 mL容量瓶中。
1.3.2 滴定法测定步骤
1.3.2.1 费林试剂的标定
用移液管吸取费林氏乙液、甲液各10mL,移入300mL三角瓶中,加入蒸馏水15mL,从滴定管加入2.5g/L标准转化糖溶液39 mL,轻轻摇匀。将三角瓶置于电炉上加热使溶液沸腾,准确煮沸2 min,加亚甲基蓝指示液3滴。在糖液保持沸腾的情况下,从滴定管继续加转化糖液,至亚甲基蓝色刚刚消失为止,即为终点。整个滴定过程溶液应保持沸腾,且滴定终点应在加入亚甲基蓝后1 min内达到。如果费林氏溶液浓度准确,则滴定耗用的2.5 g/L标准转化糖液恰好为40 mL,否则,应按以下公式计算其浓度校正系数:

式中:K——斐林试剂校正系数;
V——滴定耗用标准转化糖液体积,mL。
1.3.2.2 样品干物质百分比测定
用玻璃棒加少量样品于阿贝折光仪的棱镜面上,立即闭合棱镜。调节使明暗分界线对准在十字线上,从标尺上读取干物质百分浓度,重复操作读取3次读数,取其算数平均值,记录读数Brix。
1.3.2.3 样液制备
称取液体糖样品35.0 g于250 mL容量瓶中,精确至0.001 g,记录实际称取重量W。用移液管吸取50.0 mL样液于500 mL容量瓶中,对样品每1 g干固物加入2 mL的50 g/L草酸钾溶液,摇匀后用蒸馏水稀释至刻度,充分摇匀,过滤即可。
1.3.2.4 预检
分别用两支10 mL移液管,先吸取费林氏乙液10 mL于三角瓶内,然后再吸甲液10 mL于乙液中,混匀。用滴定管装满样液,调至刻度,从滴定管预加入样液约10mL于三角瓶内,再加入蒸馏水15 mL,摇匀,放在电炉上加热,并准确煮沸2 min。加入亚甲基蓝指示液3滴~4滴,继续滴加糖液至蓝色消失为止,即为终点,整个滴定至终点的过程不超过1 min。记录滴定耗用配制糖液毫升数V,计算复检中所需加水量等于75 mL减去配制糖液耗用量与费林试剂量20 mL。
1.3.2.5 复检
按上述次序吸取费林氏乙液、甲液各10 mL于三角瓶内,加入预检时测得的加水量,用滴定管装满样液,调至刻度,从滴定管加入比预检耗用量约少1 mL的配制样液,摇匀。滴定操作和预检相同。滴定时轻轻摇动三角瓶,但不可离开热源,使溶液继续保持沸腾,以免空气进入瓶内使亚甲基蓝被氧化而产生误差。
1.3.2.6 旋光仪测定样品蔗糖含量
取液体糖样品,将其配制成质量浓度为26.0%的待测液100 mL,将待测液倒入旋光仪中,待仪器出数,重复操作读取3次读数,取其算数平均值,记录样品蔗糖含量S。
1.3.2.7 计算及结果表示
配制糖液中含蔗糖量G计算公式如下,单位为g:式中:W——称取样品质量,g;

V——滴定耗用配制糖液,mL;
S——样品蔗糖含量,%;
Brix——液体糖试样的百分比浓度,%。液体糖样品中干物质还原糖含量计算公式如下,以%表示:
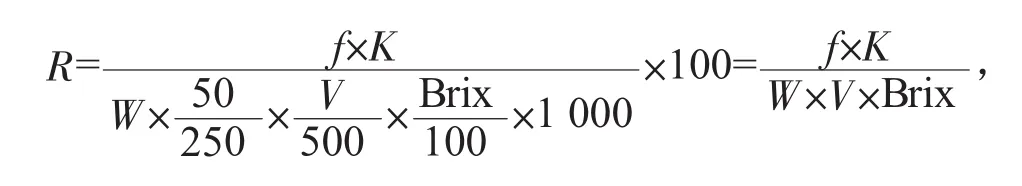
式中:R——液体糖试样还原糖的干物质含量,%;
f——校正系数(由配制糖液中含蔗糖量G查表1得);
K——费林氏溶液浓度校正系数。

表1 兰-艾农恒容法测定还原糖校正系数表
1.3.3 高效液相色谱法测定步骤
1.3.3.1 流动相的配制
超纯水:蒸馏水经过超纯水仪纯化,然后用孔径0.2 μm滤膜过滤,再经过超声波脱气即可。
1.3.3.2 标准溶液的配制
标准储备液:准确称取果糖和葡萄糖标准物质各10 g,精确至0.000 1 g,用超纯水溶解并定容至100 mL。
标准工作液:用超纯水稀释上述储备液至2.5 g/L、5 g/L、10 g/L、25 g/L、50 g/L。
1.3.3.3 绘制标准曲线
设置液相色谱工作参数:泵流速为0.6 mL/min,柱温为80℃,检测器温度为40℃,灵敏度为16。用0.22 μm的针头式过滤器及注射器依次吸取不同梯度浓度的工作液,注入进样器,分别制作果糖及葡萄糖的标准曲线,标准曲线的相关系数≥0.999。
1.3.3.4 待测液的制备与检测
用玻璃棒蘸取少量液体糖样品于阿贝折光仪的棱镜面上,立即闭合棱镜。调节使明暗分界线对准在十字线上,从标尺上读取干物质百分比浓度,重复操作读取3次读数,取其算数平均值,记录读数Brix。
准确称量2.5 g液体糖样品,记实际称取重量为m,用超纯水定容至50 mL,制得待测液。用0.22 μm的针头式过滤器及注射器吸取待测液,注入进样器检测。重复操作读取3次读数,取其算数平均值,分别记录读数为C1和C2,单位为g/L。
1.3.3.5 计算及结果表示
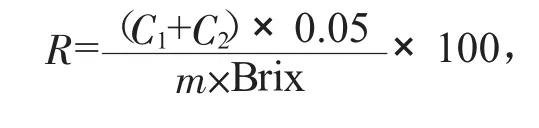
式中:R——液体糖中还原糖的干物质含量,%;
C1——液相色谱测得葡萄糖分结果,g/L;
C2——液相色谱测得果糖分结果,g/L;
0.05——定容体积,L;
m——称取的样品质量,g;
Brix——液体糖试样百分比浓度,%。
1.3.4 离子色谱仪测定步骤
1.3.4.1 流动相的配制
准确称取4.8 g的氢氧化钠于烧杯中,精确至0.001 g,用一定量超纯水溶解后转移至1 000 mL的容量瓶中,加入超纯水定容至刻度,摇匀,经0.45 μm的滤膜抽滤,待用。
1.3.4.2 标准溶液的配制
5 g/L标准溶液:准确称取果糖和葡萄糖标准物质各0.5 g,精确至0.000 1 g,用纯水溶解后转移至100 mL的容量瓶中,加入超纯水定容至刻度,摇匀,待用。
100 mg/L标准溶液:准确吸取5 g/L标准溶液2 mL,用超纯水定容至100 mL,摇匀,待用。
标准工作液:分别移取100 mg/L标准溶液50 μL、100 μL、250 μL、500 μL、1 000 μL 于 50 mL容量瓶中,用超纯水定容至刻度,摇匀,经孔径0.22 μm的滤膜过滤,待用;该系列标准工作液质量浓度分别为0.1 mg/L、0.2 mg/L、0.5 mg/L、1.0 mg/L、2.0 mg/L。
1.3.4.3 绘制标准曲线
设置离子色谱仪工作参数:泵流速1.0 mL/min,柱温30℃,进样量20 μL,分析时间为35 min。将上述梯度浓度的工作液转移至样品管中,放入自动进样器进行样品分析,根据所得数据绘制标准曲线,标准曲线相关系数≥0.999。
1.3.4.4 待测液的制备与检测
用玻璃棒蘸取少量液体糖样品于阿贝折光仪的棱镜面上,立即闭合棱镜。调节使明暗分界线对准在十字线上,从标尺上读取干物质百分比浓度,重复操作读取3次读数,取其算数平均值,记录读数Brix。
准确称取液体糖样品(0.1±0.02) g,用超纯水定容至50 mL,摇匀,经孔径0.22 μm的滤膜过滤至样品管中,放入自动进样器进行样品分析。重复操作读取3次读数,取其算数平均值,分别记录读数为C3和C4,单位为mg/L。
1.3.4.5 计算及结果表示

式中:R——液体糖试样还原糖的干物质含量,%;
C3——离子色谱测得葡萄糖分结果,mg/L;
C4——离子色谱测得果糖分结果,mg/L;
Brix——液体糖试样百分比浓度,%。
2 结果与分析
2.1 滴定法测定还原糖含量的结果
按照上述的滴定法检测步骤,对液体糖试样连续进行9组平行滴定,检测及计算结果如表2所示。由表2数据分析可知,9次平行试验滴定结果的相对平均偏差为9.02%,滴定试验结果符合QB/T 2343.2—2013中规定的相对平均偏差≤15%的要求。然而,滴定法的操作步骤繁多,造成操作误差的几率更大,因此重复滴定3次以上取其结果的平均值会更接近试样还原糖干物质含量的真实值。
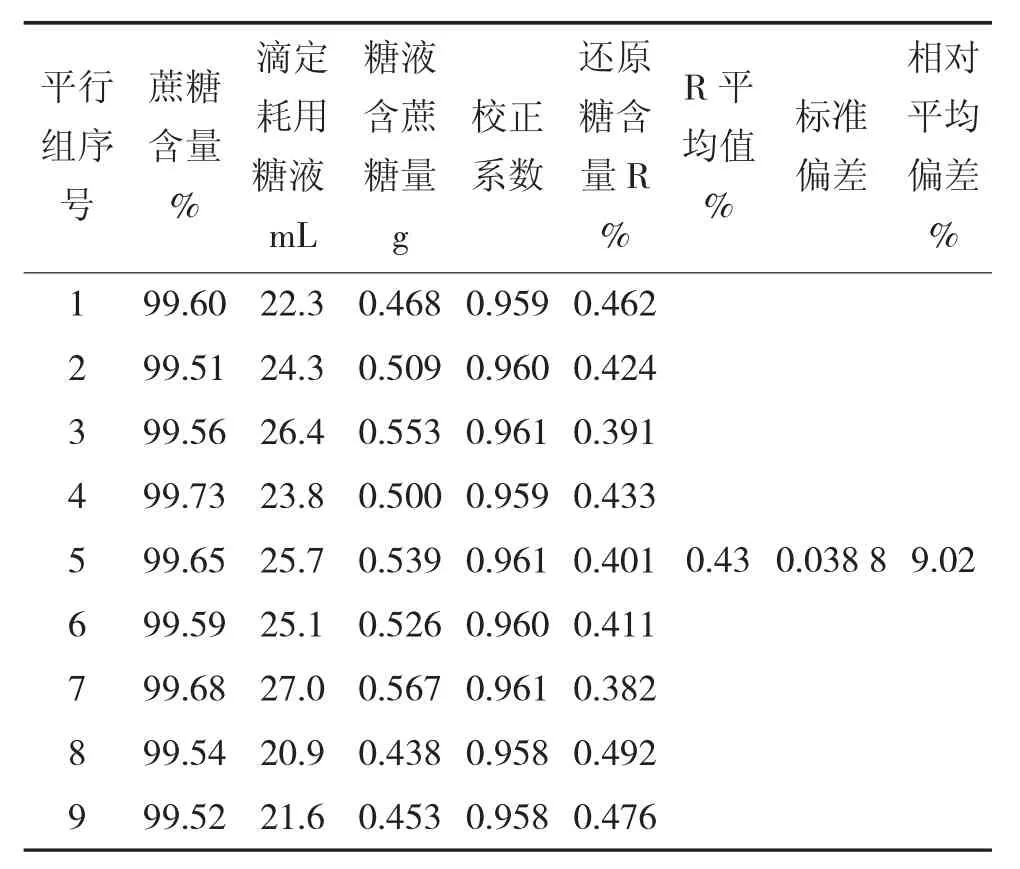
表2 兰-艾农恒容法测定结果
2.2 高效液相色谱法测定还原糖含量的结果
按照1.3.3的测定步骤,高效液相色谱法的测定图谱如图1所示。

图1 高效液相色谱法检测图谱
从高效色谱法测定图谱来看,可以清晰分辨出蔗糖峰,但是其他糖分的峰几乎难以判断。另外,糖分的结果需要判断色谱峰起点及终点位置,最终由系统自动计算出,而还原糖含量只能通过蔗糖含量间接算出。但不同的操作人对色谱峰的起点、终点位置的判断会存在一定差异,这个差异会造成>0.5%的相对误差,这个误差严重影响测定结果。在限定同一操作人的前提下,重复注入待测液进行平行测定以验证测定结果的重复性,平行测定结果如表3所示。

表3 高效液相色谱法测定结果
用高效液相色谱对液体糖试样进行9次平行测定,测定所得转化糖的平均值为0.37%,相对平均偏差为43.04%,远高于滴定法测定结果的相对平均偏差。另外从测定结果的散点分布图(图2) 可看出,9次平行测定的结果与平均值出现较大的偏移。由此可见,除了峰起点、终点判断引起的系统误差外,仪器测量本身也会存在一定的相对误差,检测方法本身存在的误差值已>0.5%,对还原糖含量极低的全蔗糖糖浆来说显然不合适。

图2 高效液相色谱法测定还原糖含量的散点分布图
2.3 离子色谱法测定还原糖含量的结果
按照1.3.4的测定步骤,离子色谱法的测定图谱如下页图3所示。
从图3可以看出,离子色谱仪能够清晰分离出全蔗糖糖浆中的葡萄糖、果糖及蔗糖等糖组分。相比起高效液相色谱,离子色谱仪检测结果的葡萄糖、果糖峰更为明显,易于判断,减少判断峰带来的系统误差。向离子色谱仪重复注入待测液进行平行测定以验证检测结果的重复性,平行测定结果如表4所示。
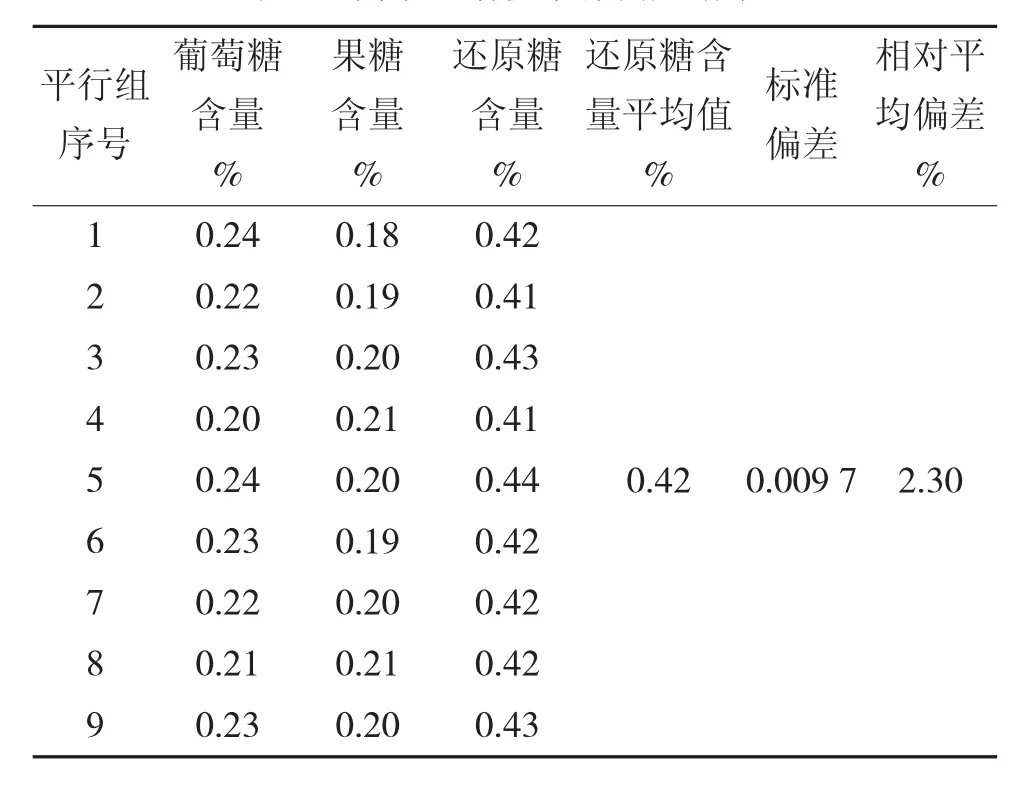
表4 离子色谱仪平行测定结果
用离子色谱仪对液体糖试样进行9次平行测定,测定所得转化糖的平均值为0.42%,与滴定法结果0.43%接近。9次平行测定的相对平均偏差为2.30%,远低于滴定法的9.02%及高效液相色谱的43.04%。从数据的准确性及重复性来看,离子色谱仪测定方法比滴定法、高效液相色谱法更适用于检测全蔗糖糖浆中的还原糖含量。

图3 离子色谱法检测图谱
3 结论
本文以液体糖(全蔗糖糖浆)为对象,研究比对了其还原糖含量的3种不同测定方法的操作差异性及其检测结果的可靠性。得出如下的结论:
1) 滴定法测定全蔗糖糖浆的操作步骤繁琐,容易引起操作误差。其测定结果的相对平均偏差在行业标准规定的控制范围内,为避免单次测量存在的偶然误差,重复测定3次以上取其结果的平均值会更接近试样还原糖干物质含量的真实值。
2) 高效液相色谱法检测步序较滴定法简单,但该方法的局限在于无法清晰识别出单糖峰,只能通过蔗糖峰间接计算出还原糖含量。而判断峰起点及终点存在的误差已经>0.5%,因此该方法用于测量全蔗糖糖浆的还原糖含量存在较大误差。
3)离子色谱仪是高效液相色谱仪的一种,其检测步骤与高效液相色谱仪类似,离子色谱仪的优势在于其检测图谱能够清晰分离出全蔗糖糖浆中的葡萄糖、果糖及蔗糖等糖组分,因而能够精确计算出全蔗糖糖浆中的还原糖含量,而且反复测定结果显示数据的重复性好,所以此方法适用于测量全蔗糖糖浆中的还原糖含量。
[1]中华人民共和国工业和信息化部.QB/T 4093—2010液体糖[S].北京:中国标准出版社,2010:1-5.
[2]吴小员,赵璧秋,黄立新.“QB/T4093—2010液体糖”旋光法测定蔗糖时还原糖的影响研究[J].甘蔗糖业,2012(6):43-47.
[3]黄彬红.返滴定法测定赤砂糖还原糖[J].福建轻纺,2014(5):46-50.
[4]雷时奋.准确认识与使用兰-艾农恒容法-对《对赤砂糖行业标准的修改意见》的一点补充[J].轻工标准与质量,1999(1):13-14.
[5]邓广华,李劲劲,农立忠,等.QB/T 4093—2010液体糖标准的问题及其修订建议[J].甘蔗糖业,2015(2):54-59.
猜你喜欢
中南民族大学学报(自然科学版)(2022年3期)2022-05-08
节能与环保(2022年3期)2022-04-26
小学生学习指导(低年级)(2020年10期)2020-11-09
广州化工(2020年6期)2020-04-18
科技视界(2019年25期)2019-11-19
中成药(2018年3期)2018-05-07
学苑创造·A版(2018年2期)2018-01-23
小猕猴智力画刊(2017年6期)2017-07-03
录井工程(2017年4期)2017-03-16
人生与伴侣·共同关注(2016年29期)2016-11-10
