
Re-evaluating the role of epithelial-mesenchymal-transition in cancer progression
2018-03-28 02:14AndrewSulaimanZeminYaoLishengWang
Andrew Sulaiman,Zemin Yao,Lisheng Wang,4,✉
1Department of Biochemistry,Microbiology and Immunology,Faculty of Medicine,University of Ottawa,Canada;
2China-Canada Centre of Research for Digestive Diseases;
3Ottawa Institute of Systems Biology,University of Ottawa,451 Smyth Road,Ottawa,Ontario K1H 8M5,Canada;
4Regenerative Medicine Program,Ottawa Hospital Research Institute,Ottawa,Ontario K1H 8L6,Canada.
Introduction
Metastasis is a process through which cancer cells dissociate from the primary tumor site,invade the surrounding tissue,hijack the circulation as a means of transport,and ultimately reconstitute the tumor at a secondary site.This process constitutes over 90%of cancer-associated deaths despite significant advances in cancer treatment[1].Epithelial-mesenchymal transition(EMT)is critical during embryo development and organogenesis.Aberrant activation of EMT is thought to promote tumor dissociation,migration,and cancer stem cell enrichment in multiple forms of cancer[2-5].These mesenchymal-like tumor cells migrate from the tumor front,through the basement membrane and into circulation where they are referred to as circulating tumor cells(CTCs)[6].A small number of CTCs display cancer stem cell(CSC)features such as immune evasion,invasiveness,tumorigenicity,and resistance to different treatments[7].Once the CSCs reach a suitable secondary tumor site,they undergo a reverse process,mesenchymal-epithelial transition(MET),halting migration and allowing reconstitution of tumor at the secondary site(Fig.1A)[10-11].
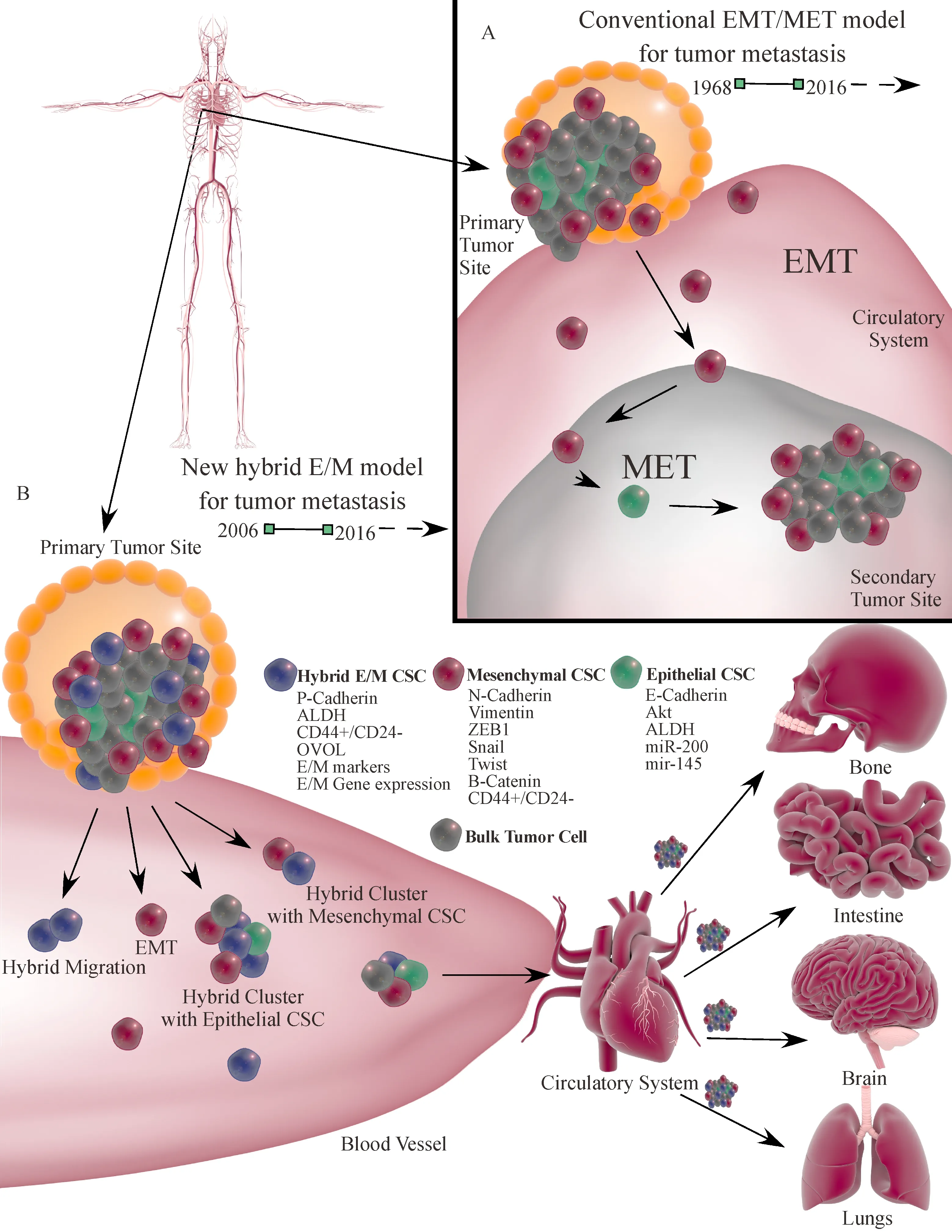
Fig.1 Schematic diagram of hybrid E/M and classical EMT/MET CSCs.(A)The classic EMT/MET model of metastasis which was coined in 1976[8].Mesenchymal cancer cells including CSCs are transformed from epithelial state through an EMT process.They then migrate outside the primary tumor,pass through the basement membrane and enter circulation.When mesenchymal CSCs reach a suitable secondary site prior to development of a new tumor,they undergo MET to regain an epithelial phenotype for tumor development.(B)The hybrid E/M theory was first succinctly formulated in 2006[9].These hybrid cancer cells including CSCs migrate from the primary tumor alone or in clusters together with epithelial or mesenchymal tumor CSCs by crossing the basement membrane to enter the circulation system and then relocate to a suitable secondary tumor site.Secondary tumor may develop from the hybrid E/M CSCs,epithelial tumor CSCs present in the cluster or mesenchymal CSCs that undergo MET.
This classic and simplified view of metastasis,during past several decades,has geared research toward targeting the migrating mesenchymal cancer cells[12-17].However,controversy has surrounded this model[18-22]which does not take into consideration of cellular plasticity,the tumorigenicity of epithelial cells,the full extent of tumor niches involved in EMT induction,the possibility of co-migration of both epithelial and mesenchymal cells,and hybrid epithelial-mesenchymal(E/M)tumor cells(Fig.1B).By addressing these deficiencies,a new model may lead to novel strategies to treat cancer metastasis and progression.
A general overview of classical EMT/MET and their regulators
The classical EMT process in cancer encompasses the gradual remodeling of epithelial-like tumor cells toward a mesenchymal-like phenotype.Mesenchymal traits include the repression of epithelial markers,enrichment of mesenchymal markers,enrichment of the CD44high/CD24lowCSC population,absence of cellular polarity due to the re-arrangement of actin cytoskeleton and redistribution of adhesion molecules,individualistic migration,and resistance to apoptosis[23-28].Epithelial traits,on the other hand,are opposite to the mesenchymal traits and exhibit some additional features such as enriched ALDH+CSC subpopulation and collective migration[29-31].
In literature,EMT is commonly characterized by decreased E-cadherin expression.E-cadherin binds to neighboring cadherins through its extracellular domain,mediating cell-cell adhesion,preventing tumor cell migration and in vivo dissemination/invasiveness[32-33].The intracellular domain of E-cadherin binds to β-Catenin(an effector of Wnt signaling),preventing the nuclear translocation of β-catenin and β-catenin/T cell factor(TCF)-mediated transactivation,impeding Wnt signaling and acquisition of mesenchymal traits[34-35].In addition to E-cadherin repression,the mesenchymal markers vimentin and N-cadherin are upregulated and EMT transcription factors(EMT-TF),such as SNAIL,SLUG,ZEB and TWIST are also upregulated.These transcription factors inhibit the epithelial phenotypes of the tumor cells while promoting acquisition of the mesenchymal phenotype through a plethora of incompletely de fined mechanisms,including microRNA networks[36],protein stabilization[37],gene expression[38],epigenetic/chromatin modi fication[39]and long noncoding RNA regulation[40].SNAIL and SLUG both inhibit E-cadherin expression,promoting β-catenin nuclear translocation and subsequent Wnt pathway upregulation[41-42].In addition,they promote the formation of the β-catenin-TCF4 transcription complex which binds to the TGF-β3 gene promoter and promoting its expression which in turn further stimulates Wnt signaling through LEF1 gene expression,ultimately enhancing acquisition of mesenchymal traits[43-44].TGF-β signaling also stimulates zinc finger E-box binding homeobox 1 and 2(ZEB1 and ZEB2)which bind to phosphorylated receptor-activated Smads[45]and various transcription factors as well as histone acetyltransferases such as p300 and p/CAF,leading to epigenetic modification of gene expression[46].Similarly,TWIST affects a large number of transcriptional processes,overrides oncogene-induced senescence and represses E-cadherin while promoting N-cadherin expression[47-48].TWIST is notably activated through hypoxia-inducible factor 1α under intratumoral hypoxic conditions[49],a trait associated with chemotherapy resistance[50].These EMT-TFs may work together through overlapping and distinct molecular mechanisms to regulate a complex network in tumor cells to control epithelial versus mesenchymal plasticity.
In addition,various biologic processes such as inflammation within the tumor microenvironment mediate EMT.When breast epithelial cells,adjacent to the tumor,were exposed to in flammatory cytokines tumor necrosis factor-α (TNF-α)and interleukin-1β (IL-1β)for 2-3 weeks,ZEB1 and SNAIL(two major EMT transcription factors)were significantly upregulated[51].The exposed breast epithelial cells then displayed upregulated matrix metalloproteinases(MMPs,capable of degrading the basement membrane to facilitate tumor cell migration)[52-53]and increased migratory/invasive capabilities,suggesting that tumor microenvironment influences plasticity and tumor cell dissemination by promoting EMT[51].
More recently,mesenchymal stem cells(MSC)from human adipose tissue have been shown to produce soluble factors after exposure to interferon-γ (IFN-γ)or TNF-α to enhance the malignancy of the MCF-7 breast cancer cells and shift the cells toward a mesenchymal phenotype with increased migration capacity,enhanced vimentin expression and decreased E-cadherin expression[54].
It has been found that bacteria can influence the tumor microenvironment and promote EMT.When gastric cancer cells were exposed to H.pylori-infected MSC supernatant enriched with IL-6(interleukin-6),IL-8 and platelet-derived growth factor-β cytokines,a mesenchymal phenotype was induced,characterized by increased migration,N-cadherin and vimentin expression while decreased E-cadherin expression[55].
Paracrine/autocrine signaling within the tumor in response to chemotherapy has also been associated with EMT promotion.IL-6,IL-8 and monocyte chemoattractant protein-1(MCP-1)cytokines along with NF-κB/IκBa and STAT3(Signal transducer and activator of transcription 3)were found to be upregulated in triple negative breast cancer cells after exposure to commonly prescribed chemotherapeutics,leading to upregulation of stem cell-associated gene and protein expression,enrichment of CD44high/CD24lowcancer stem-like cells,and enhanced tumorigenicity in nude mice[56].
Together,identification of signaling pathways and factors capable of regulating EMT has been the focus of considerable research during the past several decades in hopes that through prevention of EMT-mediated migration,tumor metastasis would have been halted[12-17].
From classical EMT/MET to the hybrid EMT/MET model
There is a plethora of literature in regards to EMT,tumor dissemination and migration through the surrounding tissue into the bloodstream and other organs.However,proof of MET at a metastatic site from a relocalized mesenchymal CTC has not yet been proved,challenging the classical EMT/MET theory regarding mesenchymal to epithelial conversion in the secondary tumor site[57].Additional arguments against classical EMT theory in metastasis and clinical applicability are the methodologies used and data generated from transgenic mice[58],xenograft implantation[59],and in vitro petri dish work[59].These experimental results are seemingly incompatible with pathological observations obtained from patients’tissues[60].Some tumors even exhibit opposite characteristics based on EMT/MET markers.For instance,in prostate cancer,secondary tumors with highly metastatic potential were found to possess a glandular appearance indicative of epithelial morphology[61].A similar phenotype is displayed in ovarian cancer which possesses elevated E-cadherin expression and an epithelial phenotype yet is highly metastatic[62-65].
To tackle the clinical applicability of EMT,lineage tracing is required.Recent reports addressed this issue by generating a mesenchymal promoter(vimentin or fibroblast specific protein-1)-induced Cre-mediated fluorescent marker in breast and lung cancer[21].The cells would irreversibly gain fluorescence in vivo upon induction of a mesenchymal phenotype through EMT.The mice spawned breast adenocarcinoma,which predominantly exhibited an epithelial phenotype based on E-cadherin expression and lacked vimentin and fluorescence expression.Lung metastasis developed spontaneously in the mouse models,which exhibited no change in fluorescence,indicating the same epithelial phenotype within the secondary tumor(con firmed via E-cadherin upregulation and vimentin repression),demonstrating that tumor cells did not activate the mesenchymal-specific promoter or undergo EMT during metastasis[21].
Additionally,another study developed a genetically engineered mouse model to delete SNAIL or TWIST through Cre-mediation in pancreatic ductal adenocarcinoma(PDAC)[22].Significantly,this deletion supressed ZEB2 and enhanced E-cadherin expression in PDAC.Lineage tracing by determining the amount of yellow fluorescent protein-tagged CTCs in the control versus SNAIL or TWIST-deletion groups showed that tumorforming potential and metastatic capacity were not affected.These results indicate that suppression of EMT-TF in PDAC mouse models did not impede tumor invasion,metastasis or dissemination when tumor cells exhibit an epithelial phenotype[22].
The aforementioned studies challenge the classical EMT model in metastasis and tumor dissemination,suggesting that EMT does not correlate with tumor dissemination and metastasis and that tumor cells with epithelial phenotypes expressing high level of E-cadherin can undergo metastasis and form secondary tumors.Although these studies use one or two core EMT related genes or EMT-TF and possibly simplify EMT processes,the findings support an incomplete,partial or hybrid EMT model to explain metastasis without losing epithelial properties and the formation of secondary tumor without interconvertible epithelial to mesenchymal transitions.
Hybrid E/M and clinical relevance
EMT is currently characterized according to the upregulated mesenchymal and repressed epithelial markers in combination with functional tests for tumor cell migration and dissemination.It is assumed that cells undergoing EMT completely switch from the epithelial to the mesenchymal phenotypes.Increasing experimental evidence,however,suggests that this switch is not a single binary decision,but rather proceeds along a spectrum,allowing for cells to express partial epithelial and mesenchymal(E/M)phenotypes and possess both E/M functionality[65-67].
Indeed,hybrid E/M states(i.e.exhibiting both epithelial and mesenchymal characteristics)have been observed in breast,brain,lung,renal,prostate and pancreatic cancers[67-72].Moreover,the hybrid E/M tumor cells display elevated CSC properties and patients show poor survival in comparison to EMT or MET phenotypes,possibly through synergy between adhesion,proliferation and migration in the E/M state[66].In breast,prostate and lung cancer patients,CTCs with E/M markers were found to migrate into blood as clusters[73-76].This collective migration would reduce anoikis and increase the chances of successful migration to a suitable secondary tumor location[77].These attributes may explain why clustered CTCs exhibit a 50-fold increase in metastatic potential[78].Hence,a better understanding of E/M properties may be key to development of an effective therapeutic strategy to control metastasis and disease relapse(Fig.1B).
Literature,however,has put the stability of the hybrid E/M tumor phenotype into question.Are these E/M tumor cells stable or is hybrid E/M tumor phenotype a fluctuating transition?Previously,E/M tumor cells were considered metastable and incapable of maintaining their E/M properties.The hybrid phenotype was thought merely a placeholder along the pathway of complete EMT or MET conversion.Recently,studies using prostate,lung and breast cancer have illustrated that this duel E/M phenotype,mediated through OVOL(OVO-like proteins)transcription,can be maintained for the extended periods of time[79-80].OVOL are a series of transcription factors(originally found through mathematical models)which play a critical role in maintaining the E/M prostate CTCs through regulation of the miR-200/ZEB and miR-34/SNAIL pathways.OVOL expression led to decreased EMT signaling induced by factors such as TGF-β,and promoted a stable shift toward the epithelial and hybrid E/M phenotype[79].
Additional mathematical modeling has identified that GRHL2 and miR-145 can also stabilize the hybrid E/M phenotype[80].Hybrid E/M lung cancer cells were able to be maintained through GRHL2,OVOL2 and miR-145 expression that act as stabilizing factors to inhibit themselves and the ZEB/miR-200 network.Knocking down miR-145 or GRHL2 led to destabilization of the E/M phenotype,driving the cells toward complete EMT induced by SNAIL[80].
Other reports have also emphasized the importance of the miR-34/SNAIL and the miR-200/ZEB regulatory networks[81].Mechanistic modeling has shown that SNAIL is able to inhibit miR-200 while ZEB is able to inhibit miR-34.As such,miR-34/SNAIL activation drives ZEB expression while inhibiting miR-200 leads to three states:high miR-200/low ZEB,low miR-200/high ZEB or medium miR-200/medium ZEB[81].These states are associated with epithelial,mesenchymal or hybrid phenotypes,respectively.E/M stabilizing factors OVOL,GRHL2 and miR-145 couple with this network,prevent ZEB signaling and promote miR-200,which inhibits complete EMT while pushing cells toward an epithelial and hybrid E/M phenotype[79-81].
Signaling pathways also affect the balance of miR-200/ZEB.For instance,NF-κB drives the LIN28/let-7 axis[82]and LIN28 inhibits let-7 which in turn inhibits ZEB[82],whereas Let-7 and miR-200 inhibit LIN-28 and bridge two networks[82].It has been found that low LIN-28 and high let-7 correlated with an epithelial phenotype while high LIN-28 mediated Let-7 inhibition and pushed cells toward a mesenchymal phenotype[82].The hybrid E/M phenotype displayed intermediate expression of LIN28 and let-7[82].Additionally,the LIN-28/let-7 axis regulates stemness through OCT4 expression[83].An outline of hybrid E/M signaling and stemness acquisition is depicted in Fig.2.
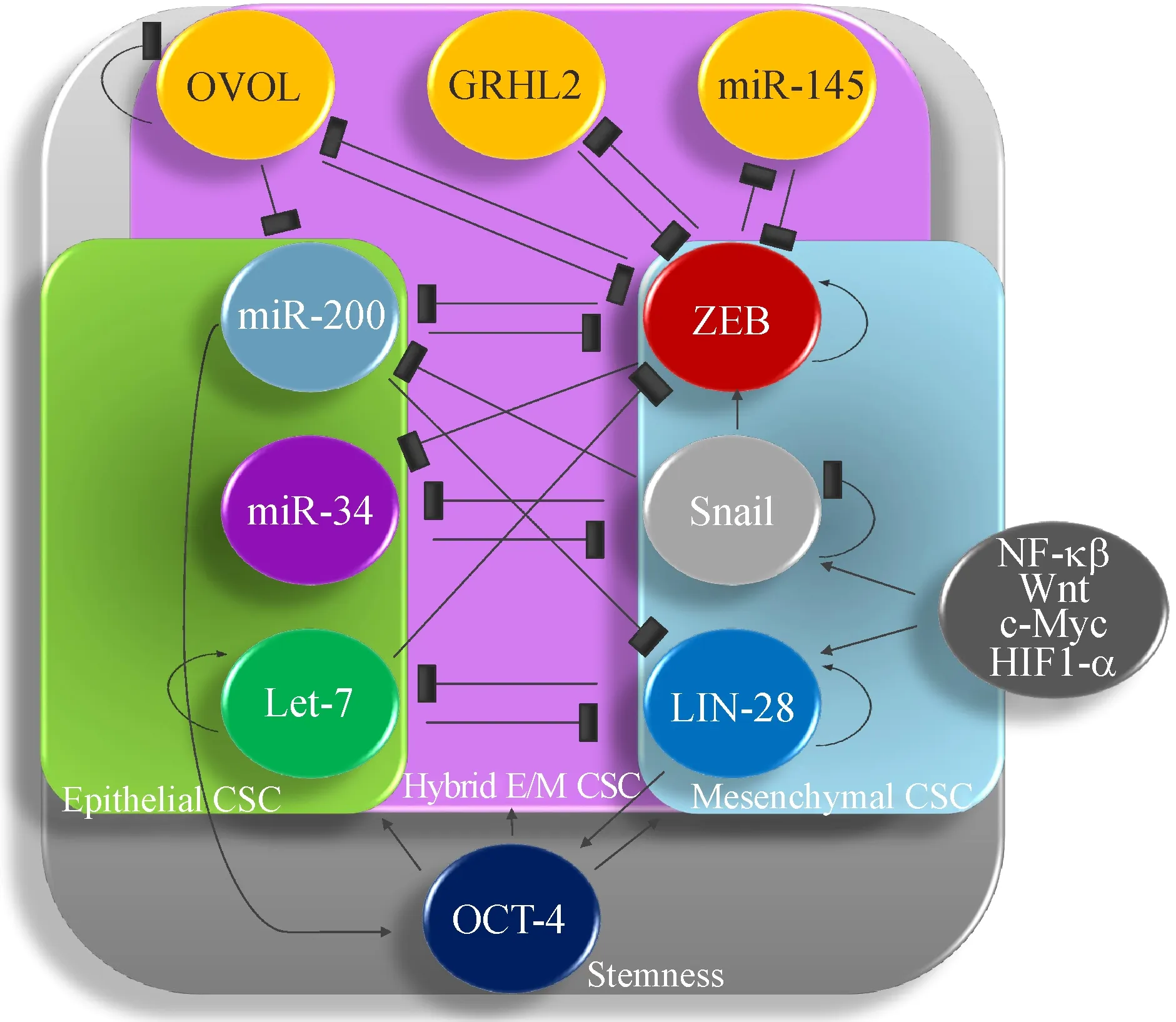
Fig.2 A schematic diagram of hybrid E/M signaling and stemness acquisition.The acquisition of mesenchymal traits is associated with increased ZEB signaling.ZEB feed-forwarding signaling inhibits miR-200 and leads to the expression of mesenchymal markers such as N-cadherin and vimentin while repressing epithelial associated markers such as E-cadherin.Snail upregulates ZEB while inhibiting miR-34.In addition,Snail is stimulated by many signaling pathways including NF-κβ,Wnt,c-Myc and HIF1-α.LIN-28 is also associated with the acquisition of mesenchymal traits.LIN-28 inhibits Let-7,increasing ZEB expression while also promoting OCT-4 and enhancing stemness.The acquisition of epithelial traits is associated with high levels of miR-200 and miR-34 which repress ZEB and Snail,respectively.MiR-200 also represses LIN-28 signaling,promoting Let-7 expression to further repress ZEB and promote OCT-4 and other stemness feature.Additionally,miR-200 inhibits LIN-28 to increase Let-7 expression,ultimately repressing mesenchymal while promoting epithelial phenotypes.The hybrid E/M phenotype is associated with intermediate signaling between miR-200/ZEB,miR-34/Snail and Let-7/LIN28 axes,which is associated with intermediate OCT-4 expression and the greatest stemness potential.OVOL,GRHL2,and miR-145 are hybrid E/M modulators,stabilizing the hybrid E/M phenotype,inhibiting ZEB signaling and complete EMT.These stabilizers also promote hybrid E/M stemness.
Further studies have shown that the acquisition of stemness can be modulated in mesenchymal,epithelial and hybrid E/M.For instance,OVOL enhanced hybrid E/M stemness while reducing mesenchymal stemness[82].On the contrary,OVOL repression exerted an opposite effect,enhancing mesenchymal while diminishing epithelial and hybrid E/M stemness[82-83].It would be interesting to determine whether or not the Wnt,Akt,YAP,and/or other signaling pathways,known in EMT/MET regulation and stemness,are involved in the miR-200/ZEB and/or LIN28/let-7 axis and associated with hybrid E/M formation,and/or involved in acquisition of stemness properties.
Investigation of hybrid E/M with improved methodologies
Studying hybrid E/M cancer cells proves to be challenging since these cells possess both epithelial and mesenchymal markers and functions.In vitro cell culture may produce inconsistent results due to artificial selection of monoculture from thriving cell sublines.Moreover,lack of microenvironment,extracellular matrix and three dimensions add to the discrepancy between in vitro and in vivo results.However,advances in the development of in vitro 3D cell culture systems have led to new discoveries in regards to cancer cell plasticity between epithelial,mesenchymal,and hybrid E/M states.
Recently,coculture of mammary EpH4 epithelial cells with a bio-engineered 3D matrix composed of solid alginate hydrogel with adhesive RGD(Arg-Gly-Asp)peptides replicated a 3D microenvironment,leading to normal epithelial morphogenesis and producing acini-like structures,native to mammary tissue[84].TGFβ1 was then used to promote EMT where mesenchymal cells were generated,but upon removal of TGFβ1,the cells switched to the hybrid E/M phenotype instead of an epithelial state.Notably,these hybrid cells displayed increased proliferative and tumorigenic capabilities and an aggressive phenotype[84].
The usage of microfluidic coculture systems for tumor microenvironment emulation has also been demonstrated to be an effective methodology for analysis of epithelial/mesenchymal/hybrid traits[85-88].This platform can analyze cancer cells in an extracellular matrix and assess proliferation,dissemination and migration in real time.Activators/repressors can be introduced into the coculture system to stimulate epithelial or mesenchymal phenotypes,and thus enable cellular communication to mimic in vivo processes.With further innovation,this system may be invaluable for further investigation of epithelial/mesenchymal and hybrid E/M characters in real time using lineage tracing with promoter-induced fluorescent proteins as described above.
Marker analysis may also be a useful tool for hybrid E/M research.Besides the dual epithelial and mesenchymal gene and protein expression,P-cadherin has been gaining traction as a hybrid E/M marker[80,89-90].P-cadherin is associated with poor prognosis in breast,oral squamous,bladder,pancreatic and ovarian cancers[91-95].It interferes with epithelial adhesion and promotes migration and metastasis through MMP upregulation,cell polarization,CDC42(cell division control protein 42 homolog)activation,and its own cleavage[89-90,96-97].Importantly,P-cadherin-promoted migration is through collective but not individual cell movement in both epithelial and mesenchymal cancer cells,mimicking the hybrid E/M phenotype[80,89-90].
CD44high/CD24low/ALDHhighmarkers may also be employed for the identification of hybrid E/M CSCs.CD44high/CD24lowsubpopulation are commonly enriched in mesenchymal-like cancer cells while ALDHhighis enriched in epithelial-like cancer cells[98-99].It has been shown in vivo in breast cancer that the ALDHhighsubpopulation resides internally while the CD44high/CD24lowtumor population lies at the tumor edge and is prone for tumor dissemination and metastasis[30].The CD44high/CD24low/ALDHhighsubpopulation in multiple breast cancer cell lines exhibited the enhanced proliferative,tumorigenic,migration,adhesive and metastatic potentials both in vitro and in vivo[30,100-102].Moreover,the CD44high/CD24low/ALDHhighsubpopulation is able to generate tumors with as few as 20 cells[101].This is consistent with the clinical data where ALDHhigh/CD44highis frequently found in patients with breast cancer and associated with increased tumor growth,disease progression,metastasis,and worsened prognosis despite radiotherapy,endocrine therapy,or chemotherapy[101,103-104].From the current literature,it seems that the CD44high/CD24low/ALDHhighmay be used for the detection of hybrid E/M CSCs.
In conclusion,while much progress has been made,targeting either epithelial or mesenchymal cancer cells seems insufficient due to cancer cell plasticity and the existence of hybrid E/M phenotype.Targeting both bulk and CSC subpopulations of epithelial,mesenchymal and hybrid E/M may be crucial for the development of clinically viable treatments to reduce resistance,relapse and metastasis.
We thank Dr.Luk Cox and Dr.Idoya Lahortiga from Somersault 18:24 to allow the use of their Library of Science and Medical Illustrations(http://www.somer-sault1824.com/resources/)for the creationof the Fig.1.
This work is supported by operating grants from Canadian Breast Cancer Foundation-Ontario Region and the Canadian Institutes of Health Research MOP-111224 to LW.
[1]Gupta GP,Massagué J.Cancer metastasis:building a framework[J].Cell,2006,127(4):679-695.
[2]Tan TZ,Miow QH,Miki Y,et al.Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients[J].EMBO Mol Med,2014,6(10):1279-1293.
[3]Garg M.Epithelial-mesenchymal transition-activating transcription factors-multifunctional regulators in cancer[J].World J Stem Cells,2013,5(4):188-195.
[4]Steinestel K,Eder S,Schrader AJ,et al.Clinical significance of epithelial-mesenchymal transition[J].Clin Transl Med,2014,3:17.
[5]Friedl P,Wolf K.Tumour-cell invasion and migration:diversity and escape mechanisms[J].Nat Rev Cancer,2003,3(5):362-374.
[6]Cristofanilli M,Hayes DF,Budd GT,et al.Circulating tumor cells:a novel prognostic factor for newly diagnosed metastatic breast cancer[J].J Clin Oncol,2005,23(7):1420-1430.
[7]Yang MH,Imrali A,Heeschen C.Circulating cancer stem cells:the importance to select[J].Chin J Cancer Res,2015,27(5):437-449.
[8]Hay ED.Organization and fine structure of epithelium and mesenchyme in the developing chick embryo[J].Epithelial-Mesenchymal Interactions,1968,2:31-35.
[9]Lee JM,Dedhar S,Kalluri R,et al.The epithelialmesenchymal transition:new insights in signaling,development,and disease[J].J Cell Biol,2006,172(7):973-981.
[10]Yao D,Dai C,Peng S.Mechanism of the mesenchymalepithelial transition and its relationship with metastatic tumor formation[J].Mol Cancer Res,2011,9(12):1608-1620.
[11]Gunasinghe NP,Wells A,Thompson EW,et al.Mesenchymal-epithelial transition(MET)as a mechanism for metastatic colonisation in breast cancer[J].Cancer Metastasis Rev,2012,31(3-4):469-478.
[12]Creighton CJ,Chang JC,Rosen JM.Epithelial-mesenchymal transition(EMT)in tumor-initiating cells and its clinical implications in breast cancer[J].J Mammary Gland Biol Neoplasia,2010,15(2):253-260.
[13]Vazquez-Martin A,Oliveras-Ferraros C,Cufí S,et al.Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition(EMT)status[J].Cell Cycle,2010,9(18):3807-3814.
[14]Davis FM,Stewart TA,Thompson EW,et al.Targeting EMT in cancer:opportunities for pharmacological intervention[J].Trends Pharmacol Sci,2014,35(9):479-488.
[15]Huang T,Chen Z,Fang L.Curcumin inhibits LPS-induced EMT through downregulation of NF-κB-Snail signaling in breast cancer cells[J].Oncol Rep,2013,29(1):117-124.
[16]Sokol JP,Neil JR,Schiemann BJ,et al.The use of cystatin C to inhibit epithelial-mesenchymal transition and morphological transformation stimulated by transforming growth factorbeta[J].Breast Cancer Res,2005,7(5):R844-R853.
[17]Tojo M,Hamashima Y,Hanyu A,et al.The ALK-5 inhibitor A-83-01 inhibits Smad signaling and epithelial-to-mesenchymal transition by transforming growth factor-beta[J].Cancer Sci,2005,96(11):791-800.
[18]Iwanami T,Uramoto H,Nakagawa M,et al.Clinical significance of epithelial-mesenchymal transition-associated markers in malignant pleural mesothelioma[J].Oncology,2014,86(2):109-116.
[19]Tarin D,Thompson EW,Newgreen DF.The fallacy of epithelial mesenchymal transition in neoplasia[J].Cancer Res,2005,65(14):5996-6000.,discussion 6000-6001.
[20]Chui MH.Insights into cancer metastasis from a clinicopathologic perspective:Epithelial-Mesenchymal Transition is not a necessary step[J].Int J Cancer,2013,132(7):1487-1495.
[21]Fischer KR,Durrans A,Lee S,et al.Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance[J].Nature,2015,527(7579):472-476.
[22]Zheng X,Carstens JL,Kim J,et al.Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer[J].Nature,2015,527(7579):525-530.
[23]Moreno-Bueno G,Portillo F,Cano A.Transcriptional regulation of cell polarity in EMT and cancer[J].Oncogene,2008,27(55):6958-6969.
[24]Lamouille S,Xu J,Derynck R.Molecular mechanisms of epithelial-mesenchymal transition[J].Nat Rev Mol Cell Biol,2014,15(3):178-196.
[25]Ma M,He M,Jiang Q,et al.MiR-487a Promotes TGF-β1-induced EMT,the Migration and Invasion of Breast Cancer Cells by Directly Targeting MAGI2[J].Int J Biol Sci,2016,12(4):397-408.
[26]Kong L,Guo S,Liu C,et al.Overexpression of SDF-1 activates the NF-κB pathway to induce epithelial to mesenchymal transition and cancer stem cell-like phenotypes of breast cancer cells[J].Int J Oncol,2016,48(3):1085-1094.
[27]Ma F,Chen D,Chen F,et al.Human Umbilical Cord Mesenchymal Stem Cells Promote Breast Cancer Metastasis by Interleukin-8-and Interleukin-6-Dependent Induction of CD44(+)/CD24(-)Cells[J].Cell Transplant,2015,24(12):2585-2599.
[28]Gonçalves NdoN,Colombo J,Lopes JR,et al.Effect of Melatonin in Epithelial Mesenchymal Transition Markers and Invasive Properties of Breast Cancer Stem Cells of Canine and Human Cell Lines[J].PLoS One,2016,11(3):e0150407.
[29]Das T,Safferling K,Rausch S,et al.A molecular mechanotransduction pathway regulates collective migration of epithelial cells[J].Nat Cell Biol,2015,17(3):276-287.
[30]Liu S,Cong Y,Wang D,et al.Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts[J].Stem Cell Reports,2013,2(1):78-91.
[31]Yauch RL,Januario T,Eberhard DA,et al.Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients[J].Clin Cancer Res,2005,11(24 Pt 1):8686-8698.
[32]Frixen UH,Behrens J,Sachs M,et al.E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells[J].J Cell Biol,1991,113(1):173-185.
[33]Vleminckx K,Vakaet LJr,Mareel M,et al.Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role[J].Cell,1991,66(1):107-119.
[34]Orsulic S,Huber O,Aberle H,et al.E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation[J].J Cell Sci,1999,112(Pt 8):1237-1245.
[35]Li Q,Mattingly RR.Restoration of E-cadherin cell-cell junctions requires both expression of E-cadherin and suppression of ERK MAP kinase activation in Ras-transformed breast epithelial cells[J].Neoplasia,2008,10(12):1444-1458.
[36]Zhang H,Li Y,Lai M.The microRNA network and tumor metastasis[J].Oncogene,2010,29(7):937-948.
[37]Díaz VM,Viñas-Castells R,García de Herreros A.Regulation of the protein stability of EMT transcription factors[J].Cell Adh Migr,2014,8(4):418-428.
[38]Minafra L,Bravatà V,Forte GI,et al.Gene expression pro filing of epithelial-mesenchymal transition in primary breast cancer cell culture[J].Anticancer Res,2014,34(5):2173-2183.
[39]Kiesslich T,Pichler M,Neureiter D.Epigenetic control of epithelial-mesenchymal-transition in human cancer.[Review][J].Mol Clin Oncol,2013,1(1):3-11.
[40]Xu Q,Deng F,Qin Y,et al.Long non-coding RNA regulation of epithelial-mesenchymal transition in cancer metastasis[J].Cell Death Dis,2016,7(6):e2254.
[41]Cano A,Pérez-Moreno MA,Rodrigo I,et al.The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression[J].Nat Cell Biol,2000,2(2):76-83.
[42]Bolós V,Peinado H,Pérez-Moreno MA,et al.The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions:a comparison with Snail and E47 repressors[J].J Cell Sci,2003,116(Pt 3):499-511.
[43]Medici D,Hay ED,Olsen BR.Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factorbeta3[J].Mol Biol Cell,2008,19(11):4875-4887.
[44]Nishita M,Hashimoto MK,Ogata S,et al.Interaction between Wnt and TGF-β signalling pathways during formation of Spemann’s organizer[J].Nature,2000,403(6771):781-785.
[45]Gregory PA,Bracken CP,Smith E,et al.An autocrine TGF-β/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition[J].Mol Biol Cell,2011,22(10):1686-1698.
[46]Postigo AA,Depp JL,Taylor JJ,et al.Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins[J].EMBO J,2003,22(10):2453-2462.
[47]Vesuna F,van Diest P,Chen JH,et al.Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer[J].Biochem Biophys Res Commun,2008,367(2):235-241.
[48]Montserrat N,Gallardo A,Escuin D,et al.Repression of E-cadherin by SNAIL,ZEB1,and TWIST in invasive ductal carcinomas of the breast:a cooperative effort[J]?Hum Pathol,2011,42(1):103-110.
[49]Yang MH,Wu MZ,Chiou SH,et al.Direct regulation of TWIST by HIF-1α promotes metastasis[J].Nat Cell Biol,2008,10(3):295-305.
[50]Song X,Liu X,Chi W,et al.Hypoxia-induced resistance to cisplatin and doxorubicin in non-small cell lung cancer is inhibited by silencing of HIF-1α gene[J].Cancer Chemother Pharmacol,2006,58(6):776-784.
[51]Leibovich-Rivkin T,Liubomirski Y,Bernstein B,et al.Inflammatory factors of the tumor microenvironment induce plasticity in nontransformed breast epithelial cells:EMT,invasion,and collapse of normally organized breast textures[J].Neoplasia,2013,15(12):1330-1346.
[52]Pei D,Weiss SJ.Transmembrane-deletion mutants of the membrane-type matrix metalloproteinase-1 process progelatinase A and express intrinsic matrix-degrading activity[J].J Biol Chem,1996,271(15):9135-9140.
[53]Bernhard EJ,Gruber SB,Muschel RJ.Direct evidence linking expression of matrix metalloproteinase 9(92-kDa gelatinase/collagenase)to the metastatic phenotype in transformed rat embryo cells[J].Proc Natl Acad Sci U S A,1994,91(10):4293-4297.
[54]Trivanović D,Jauković A,Krstić J,et al.Inflammatory cytokines prime adipose tissue mesenchymal stem cells to enhance malignancy of MCF-7 breast cancer cells via transforming growth factor-β1[J].IUBMB Life,2016,68(3):190-200.
[55]Zhang Q,Ding J,Liu J,et al.Helicobacter pylori-infected MSCs acquire a pro-inflammatory phenotype and induce human gastric cancer migration by promoting EMT in gastric cancer cells[J].Oncol Lett,2016,11(1):449-457.
[56]Jia D,Tan Y,Liu H,et al.Cardamonin reduces chemotherapyenriched breast cancer stem-like cells in vitro and in vivo[J].Oncotarget,2016,7(1):771-785.
[57]Bastid J.EMT in carcinoma progression and dissemination:facts,unanswered questions,and clinical considerations[J].Cancer Metastasis Rev,2012,31(1-2):277-283.
[58]Terao M,Ishikawa A,Nakahara S,et al.Enhanced epithelial-mesenchymal transition-like phenotype in N-acetylglucosa-minyltransferase V transgenic mouse skin promotes wound healing[J].J Biol Chem,2011,286(32):28303-28311.
[59]Yoshida T,Ozawa Y,Kimura T,et al.Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial-mesenchymal transition(EMT)to mesenchymal-epithelial transition(MET)states[J].Br J Cancer,2014,110(6):1497-1505.
[60]Ledford H.Cancer theory faces doubts[J].Nature,2011,472(7343):273-273.
[61]Rubin MA,Putzi M,Mucci N,et al.Rapid(“warm”)autopsy study for procurement of metastatic prostate cancer[J].Clin Cancer Res,2000,6(3):1038-1045.
[62]Scotton CJ,Wilson JL,Milliken D,et al.Epithelial cancer cell migration:a role for chemokine receptors[J]?Cancer Res,2001,61(13):4961-4965.
[63]Park CS,Kim TK,Kim HG,et al.Therapeutic targeting of tetraspanin8 in epithelial ovarian cancer invasion and metastasis[J].Oncogene,2016,35(34):4540-4548.
[64]Yang Y,Jiang Y,Wan Y,et al.UCA1 functions as a competing endogenous RNA to suppress epithelial ovarian cancer metastasis[J].Tumour Biol,2016,37(8):10633-10641.
[65]Christiansen JJ,Rajasekaran AK.Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis[J].Cancer Res,2006,66(17):8319-8326.
[66]Jolly MK,Boareto M,Huang B,et al.Implications of the hybrid epithelial/mesenchymal phenotype in metastasis[J].Front Oncol,2015,5:155.
[67]Rhim AD,Mirek ET,Aiello NM,et al.EMT and dissemination precede pancreatic tumor formation[J].Cell,2012,148(1-2):349-361.
[68]Grosse-Wilde A,Fouquier d’Hérouël A,McIntosh E,et al.Stemness of the hybrid epithelial/mesenchymal state in breast cancer and its association with poor survival[J].PLoS One,2015,10(5):e0126522.
[69]Andriani F,Bertolini G,Facchinetti F,et al.Conversion to stem-cell state in response to microenvironmental cues is regulated by balance between epithelial and mesenchymal features in lung cancer cells[J].Mol Oncol,2016,10(2):253-271.
[70]Jeevan DS,Cooper JB,Braun A,et al.Molecular pathways mediating metastases to the brain via epithelial-to-mesenchymal transition:Genes,proteins,and functional analysis[J].Anticancer Res,2016,36(2):523-532.
[71]Sampson VB,David JM,Puig I,et al.Wilms’tumor protein induces an epithelial-mesenchymal hybrid differentiation state in clear cell renal cell carcinoma[J].PLoS One,2014,9(7):e102041.
[72]Ruscetti M,Quach B,Dadashian EL,et al.Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis[J].Cancer Res,2015,75(13):2749-2759.
[73]Jansson S,Bendahl PO,Larsson AM,et al.Prognostic impact of circulating tumor cell apoptosis and clusters in serial blood samples from patients with metastatic breast cancer in a prospective observational cohort[J].BMC Cancer,2016,16(1):433.
[74]Danila DC,Heller G,Gignac GA,et al.Circulating tumor cell number and prognosis in progressive castration-resistant prostate cancer[J].Clin Cancer Res,2007,13(23):7053-7058.
[75]Yoon SO,Kim YT,Jung KC,et al.TTF-1 mRNA-positive circulating tumor cells in the peripheral blood predict poor prognosis in surgically resected non-small cell lung cancer patients[J].Lung Cancer,2011,71(2):209-216.
[76]Zhang L,Ridgway LD,Wetzel MD,et al.The identification and characterization of breast cancer CTCs competent for brain metastasis[J].Sci Transl Med,2013,5(180):180ra48-180ra48.
[77]Fu A,Ma S,Wei N,et al.High expression of MnSOD promotes survival of circulating breast cancer cells and increases their resistance to doxorubicin[J].Oncotarget,2016,7(31):50257-50275.
[78]Aceto N,Bardia A,Miyamoto DT,et al.Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis[J].Cell,2014,158(5):1110-1122.
[79]Jia D,Jolly MK,Boareto M,et al.OVOL guides the epithelial-hybrid-mesenchymal transition[J].Oncotarget,2015,6(17):15436-15448.
[80]Jolly MK,Tripathi SC,Jia D,et al.Stability of the hybrid epithelial/mesenchymal phenotype[J].Oncotarget,2016,7(19):27067-27084.
[81]Lu M,Jolly MK,Levine H,et al.MicroRNA-based regulation of epithelial-hybrid-mesenchymal fate determination[J].Proc Natl Acad Sci U S A,2013,110(45):18144-18149.
[82]Jolly MK,Jia D,Boareto M,et al.Coupling the modules of EMT and stemness:A tunable ‘stemness window’model[J].Oncotarget,2015,6(28):25161-25174.
[83]Jolly MK,Huang B,Lu M,et al.Towards elucidating the connection between epithelial-mesenchymal transitions and stemness[J].J R Soc Interface,2014,11(101):20140962.
[84]Bidarra SJ,Oliveira P,Rocha S,et al.A 3D in vitro model to explore the inter-conversion between epithelial and mesenchymal states during EMT and its reversion[J].Sci Rep,2016,6:27072-27072.
[85]Xu Z,Li E,Guo Z,et al.Design and Construction of a Multi-Organ Micro fluidic Chip Mimicking the in vivo Microenvironment of Lung Cancer Metastasis[J].ACS Appl Mater Interfaces,2016,8(39):25840-25847.
[86]Kim TH,Yoon HJ,Nagrath S.Identification and characterization of EMT/MET signatures in circulating tumor cells isolated from patients with metastatic breast cancer using graphene oxide nano-chip[J].Cancer Res,2016,76(14Supplement):1685-1685.
[87]Wang Y.Mutation and gene expression analysis of circulating tumor cells(CTCs)enriched and retrieved by a sensitive microfluidic device[J].Cancer Res,2016,76(14Supplement):1688-1688.
[88]Aref AR,Huang RYJ,Yu W,et al.Screening therapeutic EMT blocking agents in a three-dimensional microenvironment[J].Integr Biol(Camb),2013,5(2):381-389.
[89]Plutoni C,Bazellieres E,Le Borgne-Rochet M,et al.P-cadherin promotes collective cell migration via a Cdc42-mediated increase in mechanical forces[J].J Cell Biol,2016,212(2):199-217.
[90]Plutoni C,Bazellières E,Gauthier-Rouvière C.P-cadherinmediated Rho GTPase regulation during collective cell migration[J].Small GTPases,2016,(just-accepted):00-00.
[91]Vieira AF,Ricardo S,Ablett MP,et al.P-cadherin is coexpressed with CD44 and CD49f and mediates stem cell properties in basal-like breast cancer[J].Stem Cells,2012,30(5):854-864.
[92]Li B,Shi H,Wang F,et al.Expression of E-,P-and NCadherin and Its Clinical Significance in Cervical Squamous Cell Carcinoma and Precancerous Lesions[J].PLoS One,2016,11(5):e0155910.
[93]Wang P,Lin SL,Zhang LH,et al.The prognostic value of P-cadherin in non-muscle-invasive bladder cancer[J].Eur J Surg Oncol,2014,40(3):255-259.
[94]Sakamoto K,Imai K,Higashi T,et al.Significance of P-cadherin overexpression and possible mechanism of its regulation in intrahepatic cholangiocarcinoma and pancreatic cancer[J].Cancer Sci,2015,106(9):1153-1162.
[95]Ko SY,Naora H.HOXA9 promotes homotypic and heterotypic cell interactions that facilitate ovarian cancer dissemination via its induction of P-cadherin[J].Mol Cancer,2014,13(1):170.
[96]Mar J,Robinson A,Neville R,et al.P-cadherin modulates signaling of multiple growth factor receptors and cellular aggressiveness in oral carcinoma cells[J].Cancer Res,2014,74(19Supplement):4409-4409.
[97]Ribeiro AS,Paredes J.P-cadherin linking breast cancer stem cells and invasion:a promising marker to identify an“intermediate/metastable”EMT state[J].Front Oncol,2015,4:371.
[98]Mi K,Xing Z.CD44(+)/CD24(-)breast cancer cells exhibit phenotypic reversion in three-dimensional self-assembling peptide RADA16 nano fiber scaffold[J].Int J Nanomedicine,2015,10:3043-3053.
[99]Paholak HJ,Stevers NO,Chen H,et al.Elimination of epithelial-like and mesenchymal-like breast cancer stem cells to inhibit metastasis following nanoparticle-mediated photothermal therapy[J].Biomaterials,2016,104:145-157.
[100]Zhang M,Rosen JM.Developmental Insights into Breast Cancer Intratumoral Heterogeneity[J].Trends Cancer,2015,1(4):242-251.
[101]Ginestier C,Hur MH,Charafe-Jauffret E,et al.ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome[J].Cell Stem Cell,2007,1(5):555-567.
[102]Croker AK,Goodale D,Chu J,et al.High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability[J].J Cell Mol Med,2009,13(8B8b):2236-2252.
[103]Qiu Y,Pu T,Guo P,et al.ALDH(+)/CD44(+)cells in breast cancer are associated with worse prognosis and poor clinical outcome[J].Exp Mol Pathol,2016,100(1):145-150.
[104]Croker AK,Allan AL.Inhibition of aldehyde dehydrogenase(ALDH)activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44+human breast cancer cells[J].Breast Cancer Res Treat,2012,133(1):75-87.
 THE JOURNAL OF BIOMEDICAL RESEARCH2018年2期
THE JOURNAL OF BIOMEDICAL RESEARCH2018年2期
- THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Legal protection of the rights of clinical trial subjects in China
- Multifunctional quantum dots and liposome complexes in drug delivery
- Evaluation of cardiolipin nanodisks as lipid replacement therapy for Barth syndrome
- Pathological significance and regulatory mechanism of lymphotoxin β receptor overexpression in T cells of patients with systemic lupus erythematosus
- A novel entry point for pedicle screw placement in the thoracic spine
- Effects of leptin on femoral fracture in rats