
第一性原理研究Mn和Cu掺杂六钛酸钾(K2Ti6O13)的电子结构和光学性质∗
2018-03-26 19:06戚玉敏陈恒利金朋路洪艳崔春翔
物理学报 2018年6期
戚玉敏 陈恒利 金朋 路洪艳 崔春翔
1)(河北工业大学材料科学与工程学院,天津 300130)
2)(淮北师范大学物理与电子信息学院,淮北 235000)
1 引 言
随着社会工业化的加速发展,环境污染和能源短缺日益加剧,成为全球各国急需解决的两大问题.自1972年Fujishima和Honda[1]发现光催化现象以来,半导体光催化技术逐渐被应用于治理环境污染[2−4]和转化利用太阳能[5−7].目前使用的光催化材料基本上是宽带隙半导体[8−10],它们只能被波长较小的紫外光激发,而紫外光仅占太阳能很少的一部分(约5%),因而这些光催化材料对太阳能的利用率极低.此外光生载流子易复合,导致其光量子效率很低.近年来,为了扩宽这些光催化材料的光谱响应范围和抑制光生载流子复合,科学家们通过实验或者理论计算设计了各种方法对它们进行改性,如金属元素掺杂[11−14]、非金属元素掺杂[15−17]、金属与非金属共掺杂[18,19]、贵金属负载[20]和半导体复合[21]等.在这些方法中,金属掺杂被认为是最有效、最简单的方法之一.
近年来,六钛酸钾(K2Ti6O13)因其较高的化学稳定性、良好的物理性能和较低的制备成本[22−24],引起了材料研究人员的极大兴趣.K2Ti6O13的晶体结构由TiO6八面体通过共棱和共面连接而成,具有沿着y轴的矩形隧道结构,其中钾离子位于隧道中间[25].这种特殊的隧道结构使K2Ti6O13具有了广泛的应用,如作为增强材料、隔热涂料、耐摩擦材料、过滤材料等[26,27],特别是作为光催化剂用于有毒物质降解和纯水分解[28,29].然而,K2Ti6O13也是宽带隙半导体,其带隙值约3.45 eV[30],因而对太阳光的利用率很低.因此,为了使K2Ti6O13对可见光响应,有必要对其进行合理改性.3d过渡金属元素由于d电子态的存在,掺入光催化材料后会对其光催化性能产生影响,特别是用于取代掺杂TiO2中和其同一周期的Ti元素[31,32].根据已有研究报道,Mn,Cu的掺杂能使TiO2实现对可见光的吸收[33,34],因此,本文研究Mn,Cu部分取代K2Ti6O13中的Ti原子对光谱响应范围的影响.本文应用密度泛函理论计算,通过3d过渡金属Mn和Cu掺杂前后六钛酸钾电子结构和光学性质的变化,探讨了Mn和Cu掺杂对六钛酸钾光吸收能力的影响及其光催化改性的内在机理,期望为将来的实验提供必要的理论基础和参考.
2 模型与计算方法
六钛酸钾属单斜晶系,空间群为C2/m[27,35],每个晶胞含有2个K2Ti6O13分子单元.如图1所示,本文计算采用K2Ti6O13(1×2×1)超胞结构模型,其中Mn,Cu分别取代了K2Ti6O13中的Ti原子.计算主要采用Materials Studio软件中的Castep模块完成[36]. 计算时首先对结构进行优化,优化时电子间的交换关联能采用广义梯度近似[37]中的PW91形式,布里渊区的积分采用2×3×3的k点设置,平面波截断能设为340 eV.优化时的收敛标准为:最大位移为1.0×10−4nm,内应力收敛标准为0.05 GPa,原子间的相互作用力最大为0.3 eV/nm,结构的总体能量收敛于1.0×10−5eV/atom.然后对电子结构和光学性质进行计算,计算中考虑的各元素价电子为Ti 3s23p63d24s2;O 2s22p4;K 3s23p64s1;Mn 3s23p63d54s2和Cu 3s23p63d104s1.此外,为了进一步分析Mn和Cu的3d轨道对杂质能级的作用,采用Quantum-ESPRESSO软件[38]对Cu和Mn的3d轨道分裂的dz2,dx2−y2,dxy,dyz和dxz轨道进行了分波态密度计算.
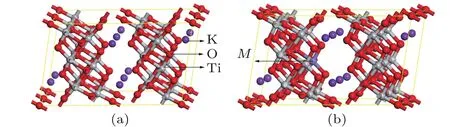
图1 (a)纯K2Ti6O13(1×2×1)超胞结构;(b)M掺杂的K2Ti6O13超晶胞结构(M=Mn或Cu)Fig.1.(a)1×2×1 supercell of pristine K2Ti6O13;(b)supercell of M-doped K2Ti6O13(M=Mn or Cu).
3 结果与讨论
3.1 晶体结构
对Cu,Mn掺杂K2Ti6O13前后的超晶胞模型几何优化后,所得晶格常数见表1.从表1可以看出,纯K2Ti6O13的计算结果与实验测试结果[35]和其他计算结果[39]都符合得很好,这说明我们的计算方法是合理的.相对于未掺杂的K2Ti6O13,Cu和Mn掺杂后K2Ti6O13晶胞体积稍有减小,这可能是由于掺杂原子的半径小于Ti原子的半径,从而造成了晶格畸变,致使键长变短.
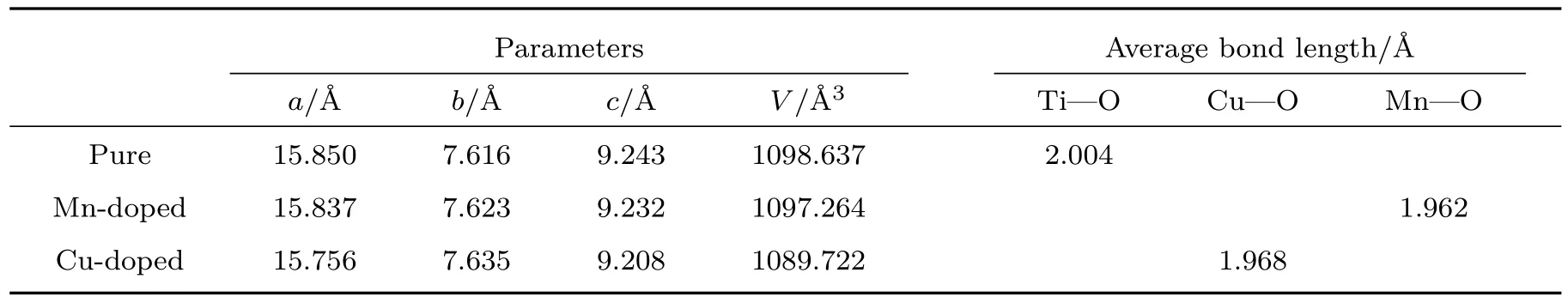
表1 几何优化后纯K2Ti6O13和Mn,Cu掺杂后K2Ti6O13的超晶胞参数Table 1.Parameters and average bond lengths of pure,Mn-doped and Cu-doped K2Ti6O13for optimized structure.
3.2 电子结构
从图2(a)可以看出,K2Ti6O13禁带宽度为2.834 eV,这和已有的计算结果相近[39,40],都小于实验值3.45 eV[30].虽然当前计算方法对带隙值有所低估,但是其相对值是有意义的,可作为一种有效的近似方法对能带和态密度等进行定性分析[41,42].
在光催化反应中,光催化材料的电子结构起着十分关键作用,它直接影响着光催化材料载流子的产生、输运、复合等行为.光催化材料带隙的大小决定着其对太阳光的响应范围,光生电子和空穴只有在能量大于或等于光催化材料带隙的光子照射到光催化材料上时才能激发产生.因而光催化材料的带隙越小,对应的吸收波长范围越大,对太阳光利用越充分.对比图2(a)和图2(b)可知,Mn掺杂后K2Ti6O13的导带和价带明显下移(导带下移1.617 eV,价带下移1.507 eV),因此带隙值减少0.11 eV,这有利于吸收带边红移.同时,Mn掺杂后,在K2Ti6O13的禁带中间出现了杂质能级,此杂质能级与价带和导带之间的能量差分别为1.407 eV和1.122 eV,可作为电子从价带跃迁至导带的桥梁.电子可先被能量较小的光子激发从价带跃迁到杂质能级,然后继续吸收光子从杂质能级跃迁到导带,因此外界只需提供较小的能量就可以将电子激发,从而实现了K2Ti6O13对可见光的吸收,提高了光的利用率.对比图2(a)和图2(c)可知,Cu掺杂的K2Ti6O13导带和价带都下移(导带下移0.42 eV,价带下移0.459 eV),其带隙值增大0.04 eV.虽然Cu掺杂后K2Ti6O13带隙略微变大,但是其杂质能级位于价带顶,与价带几乎相连,如果对带隙的评估考虑杂质能级,其带隙值将大大减小到只有1.886 eV,从而实现K2Ti6O13对可见光的响应.此外光生载流子易复合,致使大多数光催化材料量子效率低.因此,抑制载流子复合是提高光催化材料光催化活性的重要途径之一.在掺杂的光催化材料中,位于导带底和价带顶附近的杂质能级可形成离子陷阱,从而抑制电子和空穴的复合,而位于禁带中心的杂质能级一般会成为电子-空穴对的复合中心.载流子的有效质量是研究载流子运动的一个重要参数,其中电子和空穴的有效质量相差越大,意味着电子空穴复合率越低[43].通过分析能带结构,电子和空穴的有效质量可由公式得出,其中k为能带极值处的波矢,E为波矢k处的能量.虽然Mn掺杂后出现的杂质能级位于禁带中间,但是计算结果表明其电子和空穴有效质量相差很大,因此其载流子的复合率较低.由图2(c)可知,Cu掺杂后的杂质能级位于价带顶附近是受主能级,可以形成离子陷阱俘获电子,减少电子空穴的复合率,从而有希望提高K2Ti6O13的光催化性能.
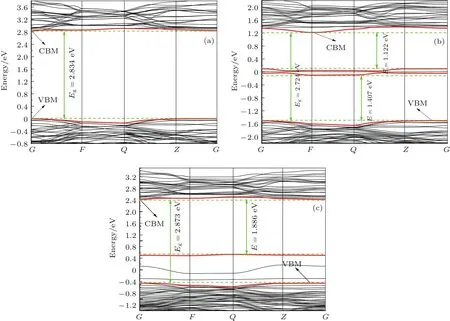
图2 (a)纯K2Ti6O13,(b)Mn掺杂K2Ti6O13和(c)Cu掺杂K2Ti6O13的能带结构Fig.2.Band structures of(a)pure K2Ti6O13,(b)Mn-doped K2Ti6O13,and(c)Cu-doped K2Ti6O13.
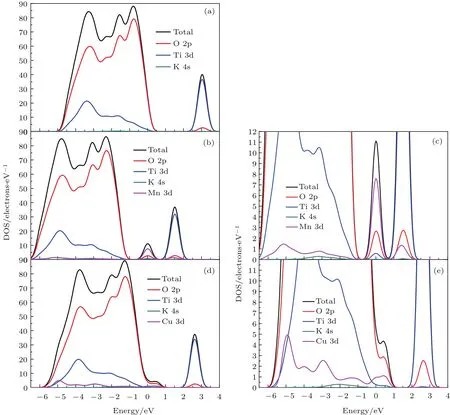
图3 (a)纯 K2Ti6O13的分态密度;(b)Mn掺杂K2Ti6O13的分态密度;(c)部分放大的Mn掺杂K2Ti6O13的分态密度;(d)Cu掺杂K2Ti6O13的分态密度;(e)部分放大的Cu掺杂K2Ti6O13的分态密度Fig.3.(a)Density of states of pure K2Ti6O13;(b)density of states of Mn-doped K2Ti6O13;(c)enlarged density of states of Mn-doped K2Ti6O13;(d)density of states of Cu-doped K2Ti6O13;(e)enlarged density of states of Cu-doped K2Ti6O13.
由图3(a)可知,掺杂前K2Ti6O13的价带和导带主要由O 2p和Ti 3d态组成,K 4s态对其几乎没有贡献,这说明Ti和O之间存在强烈的相互作用.Ti 3d和O 2p之间的相互作用决定了K2Ti6O13晶体的结构稳定性[40].由图3(b)和图3(c)可知,Mn掺杂后在K2Ti6O13的禁带中间出现了一个小的态密度峰,这个态密度峰和图2(b)中的杂质能级相对应,这说明能带中的杂质能级由O 2p,Ti 3d和Mn 3d轨道杂化而成.由图3(d)和图3(e)可知,Cu掺杂后在K2Ti6O13的价带边缘出现了一个小的态密度峰,这与图2(c)中的杂质能级相对应,说明能带中的杂质能级由O 2p,Ti 3d和Cu 3d轨道杂化而成.为了进一步分析杂质能级形成的物理机理,采用Quantum-ESPRESSO软件对Cu和Mn的3d轨道的dz2,dx2−y2,dxy,dyz和dxz轨道分波态密度进行了计算,见图4.对比图4(a)和图3(b),图4(c)和图3(d),两种计算方法得到的态密度中杂质能级的位置基本相符,这证明了我们的计算是合理的.由图3(c)和图4(b)可知,Mn掺杂后能带中间的杂质能级主要是由Mn 3d轨道的五个分裂轨道共同作用.由图3(e)和图4(d)可知,除了O 2p轨道的贡献外,Cu掺杂后价带顶附近的杂质能级主要由Cu 3d轨道分裂的dzx,dx2−y2和dz2轨道的作用.总而言之,由于Cu 3d和Mn 3d轨道的作用使掺杂后的K2Ti6O13能带中出现了杂质能级进而将提高K2Ti6O13的光催化性能,正如已有的研究指出,过渡金属的掺杂增强光催化材料的光催化活性源于其3d轨道的作用[44].
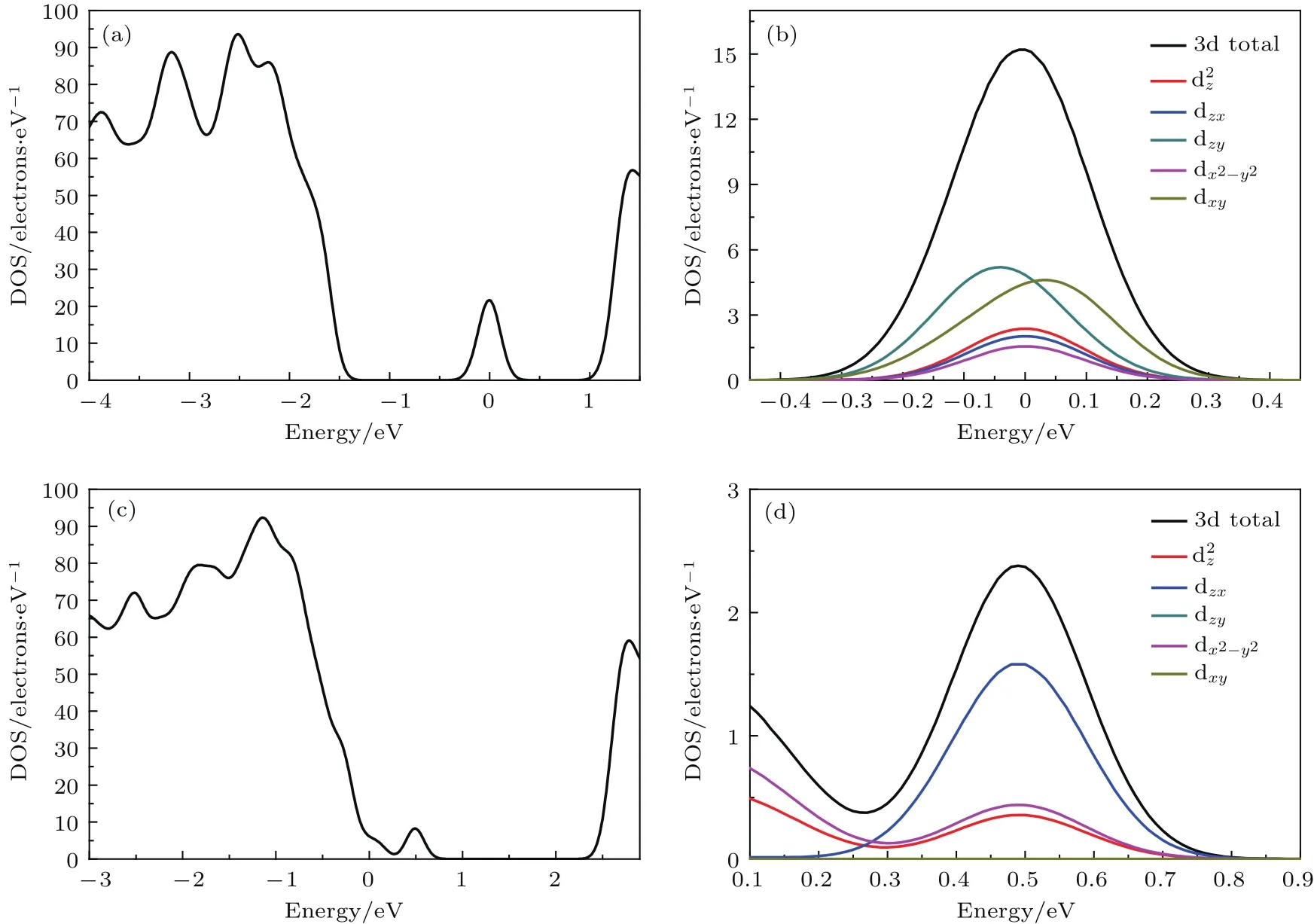
图4 (a)Mn掺杂K2Ti6O13的总态密度;(b)Mn掺杂K2Ti6O13的Mn 3d轨道的分态密度;(c)Cu掺杂K2Ti6O13的总态密度;(d)Cu掺杂K2Ti6O13的Cu 3d轨道分态密度Fig.4.(a)The total density of states of Mn-doped K2Ti6O13;(b)the partial density of states of 3d orbits of Mn;(c)the total density of states of Cu-doped K2Ti6O13;(d)the partial density of states of 3d orbits of Cu.
3.3 光学性质
在CASTEP中,光学性质是在能带结构的基础上依照电偶极子近似来进行计算.根据费米黄金定则,由占据态和非占据态波函数之间的动量矩阵元可推出介电函数的虚部ε2,即[45]
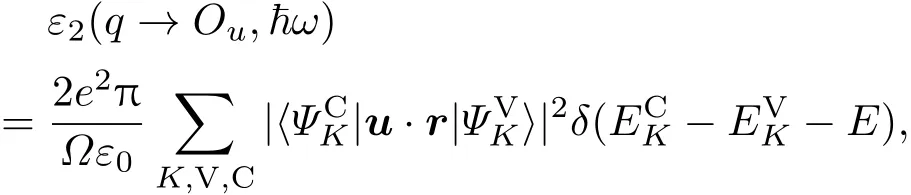
其中,u为入射电场的极化方向向量;K为倒格矢;C,V表示导带和价带;分别为价带和导带上的本征能级;〉为动量跃迁矩阵.介电函数的实部ε1可以根据ε1和ε2的Kramer-Kronig色散关系求出,此外从介电函数的虚部出发可以推导出吸收率和反射率等光学常数.由于密度泛函理论计算的带隙偏小,为了使计算结果更符合实际情况,本文在计算光学性质时使用剪刀算符方法进行修正,修正值取0.616 eV(纯K2Ti6O13禁带宽度实验测量值3.45 eV,与计算值2.834 eV之差).
介电函数虚部ε2是能带结构的直接表现形式.其峰值与通过光子激发从占据状态到未占据状态的电子跃迁过程有关,各个峰代表着不同的带间跃迁.图5所示为纯K2Ti6O13与Mn,Cu单掺杂K2Ti6O13的介电函数虚部随光子能量的变化.在高能方向Mn和Cu单掺杂后K2Ti6O13的介电函数虚部值几乎未变,这是由于掺杂前后K2Ti6O13的带隙值很接近;在4.5 eV附近,它们都有较大的峰值,这源于价带上的电子到导带的跃迁;Mn掺杂的K2Ti6O13在4.0 eV附近有个较小的峰值,对比图3(a)、图3(c)和图3(e)可知,这个峰值源于价带中的电子到导带中Mn 3d轨道的跃迁.但在低能方向掺杂后的K2Ti6O13介电函数发生明显红移,纯K2Ti6O13的介电函数值在能量小于3.5 eV时基本为零.Mn掺杂的K2Ti6O13在能量值为2—3 eV时出现了一个峰值,这是因为电子在吸收能量发生跃迁的过程中,能带中间的杂质能级起到了桥梁的作用,降低了发生跃迁所需要的能量.对于Cu掺杂的K2Ti6O13,在能量值为1—2.5 eV时出现了多个峰值,这是因为杂质能级与价带相连,可看成与O 2p,Ti 3d轨道复合成价带顶,使能隙值降低,进而降低了电子跃迁所需要的能量,这些峰值源于杂质能级上的电子到导带的跃迁.
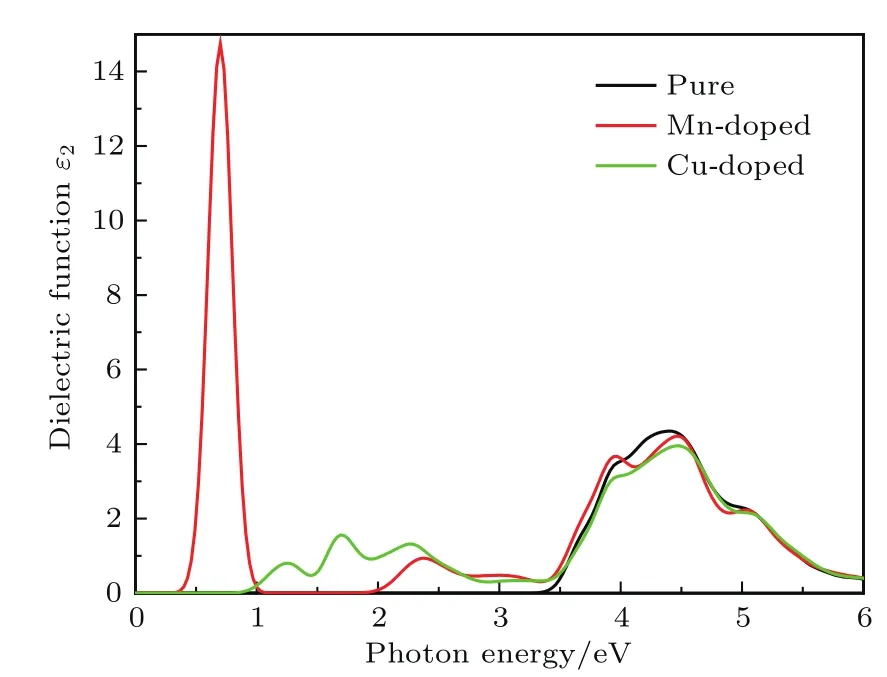
图5 纯K2Ti6O13,Mn掺杂K2Ti6O13和Cu掺杂K2Ti6O13的介电函数虚部随光子能量的变化Fig.5.Imaginary part of dielectric function as a function of photon energy for pure K2Ti6O13,Mn-doped K2Ti6O13and Cu-doped K2Ti6O13..
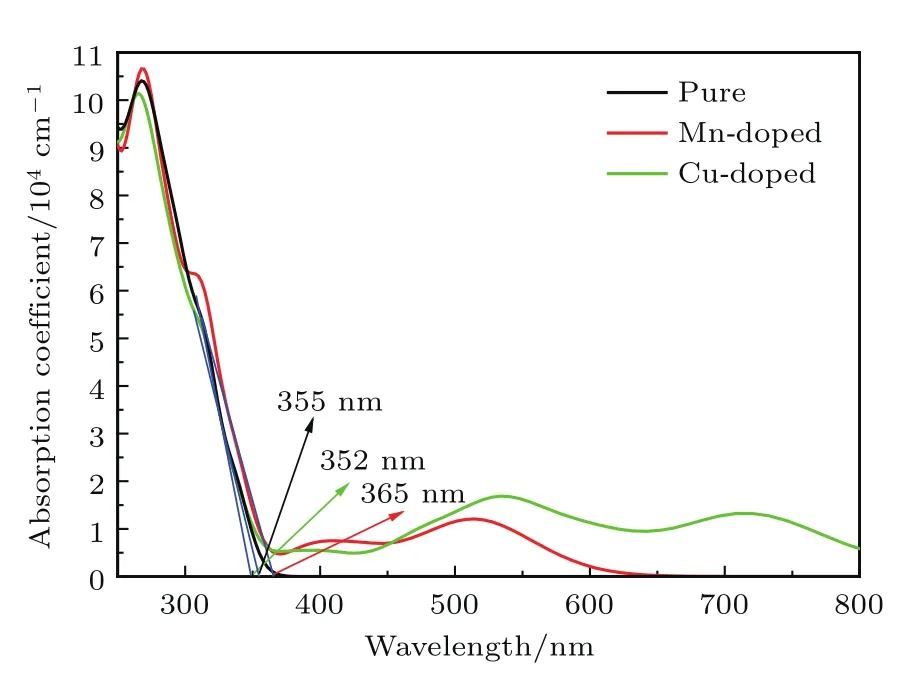
图6 纯K2Ti6O13,Mn掺杂K2Ti6O13和Cu掺杂K2Ti6O13的吸收光谱Fig.6.Absorption spectra of pure K2Ti6O13,Mndoped K2Ti6O13and Cu-doped K2Ti6O13.
由图6可知,计算所得的纯K2Ti6O13的吸收范围约在0—400 nm,这和已有的实验研究结果相符[30,46],说明对光学性质计算进行“剪刀算符”修正是必要的,也再次证明了我们所采用的计算方法是合理的.纯K2Ti6O13,Mn掺杂和Cu掺杂K2Ti6O13的吸收边分别约为355,365和352 nm,Mn掺杂后K2Ti6O13的吸收边略有红移,Cu掺杂后K2Ti6O13吸收边几乎未变,这与其理论计算的带隙值变化趋势相符.此外对于Mn掺杂的K2Ti6O13在波长为300—310 nm范围里吸收强度下降趋势变缓,这和介电函数虚部在4.0 eV附近出现较小的峰值相对应,即源于导带中Mn 3d轨道的作用.Mn和Cu的掺杂都使K2Ti6O13吸收范围向可见光区拓展,其中Mn掺杂使其吸收范围扩展到约400—650 nm,Cu掺杂K2Ti6O13的吸收范围约在400—800 nm.对照上文对电子结构变化的分析可以看出,掺杂后体系的光学性质的变化是与其电子结构的变化对应的.值得一提的是,Cu掺杂改性的K2Ti6O13,无论是吸收强度还是吸收范围都远好于Mn掺杂改性的K2Ti6O13,这可能是由于其杂质能级既可降低带隙的宽度又可提高光生载流子的效率.
4 结 论
本文采用第一性原理计算研究了3d过渡金属Mn和Cu掺杂K2Ti6O13的能带结构、电子态密度、介电函数虚部和吸收光谱.计算结果表明,Mn掺杂后K2Ti6O13带隙值变小且在能带中间出现了杂质能级,杂质能级可作为电子跃迁的桥梁,进而实现了对可见光的吸收.Cu掺杂K2Ti6O13的带隙值虽然有所增加,但是其杂质能级与价带顶相连,如果考虑杂质能级,其禁带宽度将大大减小.同时此杂质能级可抑制光生载流子的复合,提高光催化的效率.因此Cu掺杂大大拓宽了K2Ti6O13对可见光的吸收范围,且其吸收强度也大大增加.期望我们的计算预测对后续K2Ti6O13光学性质改性实验研究提供理论借鉴.
[1]Fujishima A,Honda K 1972Nature238 37
[2]Su J,Lin Z,Chen G 2016Appl.Catal.B:Environ.186 127
[3]Li C,Chen G,Sun J,Rao J,Han Z,Hu Y,Xing W,Zhang C 2016Appl.Catal.B:Environ.188 39
[4]Lou S,Jia X,Wang Y,Zhou S 2015Appl.Catal.B:Environ.176 586
[5]He Y R,Yan F F,Yu H Q,Yuan S J,Tong Z H,Sheng G P 2014Appl.Energ.113 164
[6]Osterloh F E 2008Chem.Mater.20 35
[7]Ran J,Zhang J,Yu J,Jaroniec M,Qiao S Z 2014Chem.Soc.Rev.43 7787
[8]Zhao Z,Liu Q 2008J.Phys.D:Appl.Phys.41 025105
[9]Tian Z,Liang C,Liu J,Zhang H,Zhang L 2011J.Mater.Chem.21 18242
[10]Li D,Haneda H 2003Chemosphere51 129
[11]Zhu J,Chen F,Zhang J,Chen H,Anpo M 2006J.Photochem.Photobiol.A:Chem.180 196
[12]Qin L Z,Liang H,Liao B,Liu A D,Wu X Y,Sun J 2013Nucl.Instrum.Meth.Phys.Res.Sect.B307 385
[13]Guo M,Du J 2012Phys.Rev.B:Condens.Matter407 1003
[14]Impellizzeri G,Scuderi V,Romano L,Sberna P M,Arcadipane E,Sanz R,Scuderi M,Nicotra G,Bayle M,Carles R 2014J.Appl.Phys.116 173507
[15]Liu G,Yang H G,Wang X,Cheng L,Pan J,Lu G Q,Cheng H M 2009J.Am.Chem.Soc.131 12868
[16]Pan J H,Zhang X,Du A J,Sun D D,Leckie J O 2008J.Am.Chem.Soc.130 11256
[17]Wang D H,Jia L,Wu X L,Lu L Q,Xu A W 2012Nanoscale4 576
[18]Zhang K,Wang X,Guo X,He T,Feng Y 2014J.Nanopart.Res.16 2246
[19]Zhang R,Wang Q,Liang J,Li Q,Dai J,Li W 2012Phys.B:Condens.Matter407 2709
[20]Anpo M,Takeuchi M 2003J.Catal.216 505
[21]Fujii H,Inata K,Ohtaki M,Eguchi K,Arai H 2001J.Mater.Sci.36 527
[22]Hakuta Y,Hayashi H,Arai K 2004J.Mater.Sci.39 4977
[23]Kapusuz D,Kalay Y E,Park J,Ozturk A 2015J.Ceram.Process.Res.16 291
[24]Li Y,Yu H,Yang Y,Zheng F,Ni H,Zhang M,Guo M 2016Ceram.Int.42 11294
[25]Xie J,Lu X,Zhu Y,Liu C,Bao N,Feng X 2003J.Mater.Sci.38 3641
[26]Murakami R,Matsui K 1996Wear201 193
[27]Han P D,Liang J,Yu Y,Bao H Q,Liu X G,Xu B S 2005Rare Metal Mat.Eng.34 56(in Chinese)[韩培德,梁建,余愿,鲍慧强,刘旭光,许并社 2005稀有金属材料与工程34 56]
[28]RamíRez-Salgado J,Djurado E,Fabry P 2004J.Eur.Ceram.Soc.24 2477
[29]Pescatori M,Quondamcarlo C 2003Chem.Phys.Lett.376 726
[30]Du G H,Chen Q,Han P D,Yu Y,Peng L M 2003Phys.Rev.B67 035323
[31]Wang Y,Zhang R,Li J,Li L,Lin S 2014Nanoscale Res.Lett.9 46
[32]Wu S X,Ma Z,Qin Y N,Qi X Z,Liang Z C 2004Acta Phys.Chim.Sin.20 138(in Chinese)[吴树新,马智,秦永宁,齐晓周,梁珍成2004物理化学学报20 138]
[33]Deng Q R,Xia X H,Guo M L,Gao Y,Shao G 2011Mater.Lett.65 2051
[34]Colón G,Maicu M,Hidalgo M C,Navío J A 2006Appl.Catal.B:Environ.67 41
[35]Andersson S,Wadsley A D 1962Acta Crystallogr.15 194
[36]Segall M D,Lindan P J D,Probert M,Pickard C J,Hasnip P J,Clark S J,Payne M C 2002J.Phys.:Condens.Matter14 2717
[37]Perdew J P,Burke K,Ernzerhof M 1996Phys.Rev.Lett.77 3865
[38]Giannozzi P,Baroni S,Bonini N,et al.2009J.Phys.:Condens.Matter21 395502
[39]Hua M,Li Y,Long C,Xia L 2012Physica B407 2811[40]Hua M Y,Li Y M,Li X 2011J.Synth.Cryst.40 1573(in Chinese)[华熳煜,李益民,李夏2011人工晶体学报40 1573]
[41]Stamp flC,Walle C G V D 1999Phys.Rev.B:Condens.Matter59 5521
[42]Perdew J P,Levy M 1983Phys.Rev.Lett.51 1884
[43]Wan H,Xu L,Huang W Q,Huang G F,He C N,Zhou J H,Peng P 2014Appl.Phys.A116 741
[44]Yang K,Li D F,Huang W Q,Xu L,Huang G F,Wen S 2017Appl.Phys.A123 96
[45]Zhao Z Y,Liu Q J,Zhu Z Q,Zhang J 2008Acta Phys.Sin.57 3760(in Chinese)[赵宗彦,柳清菊,朱忠其,张瑾2008物理学报57 3760]
[46]Li J,Zhang Y C,Zhang M 2012Mater.Lett.79 136
猜你喜欢
装备维修技术(2021年36期)2021-10-25
小天使·聪聪画刊(2021年2期)2021-09-10
弹箭与制导学报(2021年3期)2021-07-30
汽车零部件(2020年10期)2020-11-09
原子与分子物理学报(2019年4期)2019-09-17
汉语世界(The World of Chinese)(2019年6期)2019-09-10
网印工业(2019年4期)2019-05-21
重型机械(2019年2期)2019-04-28
西安工业大学学报(2019年2期)2019-04-02
吉首大学学报(自然科学版)(2018年3期)2018-07-03
