
气相色谱-质谱法测定染整助剂中19种有害有机溶剂
2018-03-06 08:29:40丁友超曹锡忠
分析测试学报 2018年2期
周 佳,汤 娟,丁友超*,曹锡忠
(1.江苏出入境检验检疫局工业产品检测中心,江苏 南京 210001;2.江苏省检验检疫科学技术研究院,江苏 南京 210001)
纺织原料在生产和初加工过程中常会使用或接触有毒有害物质。对纺织产品中有害物质的“源头控制”,就是对纺织原料和纺织染整助剂中的有害物质进行检测、监控。在有机反应中,有机溶剂不但广泛用作反应的溶剂,也是有机合成的重要中间体。有机溶剂的广泛应用,导致其在染整助剂及染料生产合成中的残留,进而危害人体健康。研究表明:乙二醇醚类吸收入人体后,将作用于富含脂类物质的神经、血液系统,以及肝肾等实质脏器,同时对皮肤和黏膜也有一定的刺激性[1-3],可对人体的血液循环系统和神经系统造成永久性损害,长期高浓度的接触还会致癌。1-甲基-2-吡咯烷酮能导致出生缺陷和其它生殖毒性,因此美国工业卫生学会(AIHA)研究认为,1-甲基-2-吡咯烷酮的工作场所环境暴露水平(WEEL)为10 ppm(8 小时(皮肤) )[4]。N,N-二甲基甲酰胺对眼睛、皮肤和呼吸道均具有一定的刺激作用,也可能引起食欲不振、恶心、呕吐或便秘等症状,严重时会导致中毒性肝病。
2012年1月,国际生态纺织工业协会新颁布的生态纺织品标准Oeko-Tex Standa100限定物质清单中增加了1项“溶剂残留(Solvent residues)”的要求,规定1-甲基-2吡咯烷酮和N,N-二甲基乙酰胺的残留量均不得超过0.1%。OEKO-TEX Standard 100列出了4种酰胺类有机溶剂,欧盟REACH法规限定的高关注物质(SVHC)清单不断更新,涉及15种有害溶剂。美国环境保护局(EPA)对乙二醇醚类物质提出要求;而有害化学品零排放(ZDHC)生产限用物质清单更是列出了12种有机溶剂。
目前,有机溶剂的检测对象主要是药物[5-7]、食品包装材料[8]、水[9-10]、空气[11]、化妆品[12-13]、纺织品[14-17]和皮革制品[18-19]等样品,染整助剂中有害溶剂的测试报道较少,且未见同时检测染整助剂中19种有害溶剂的研究报道。染整助剂基质比纺织品复杂,本研究采取超声萃取技术提取染整助剂中的有机溶剂,萃取后进气相色谱-质谱仪分析,建立了同时测定染整助剂中19种有机溶剂残留量的气相色谱-质谱联用分析方法。
1 实验部分
1.1 仪器、试剂与材料
Thermo Trace ISQ气相色谱-质谱联用仪(美国Thermo公司);KQ-500DE超声波发生器(德国Barnstead公司);Legend Micro 17型离心机(美国Thermo公司),管状玻璃反应器(约15 mL),0.22 μm聚四氟乙烯薄膜滤头(津腾公司),0.22 μm有机相针式滤器(上海安谱科学仪器有限公司)。
色谱纯甲醇(美国Media公司),分析纯试剂甲醇、乙酸乙酯、丙酮、二氯甲烷(南京化学试剂公司)。
标准品乙二醇二甲醚(纯度99.5%)、乙二醇二乙醚(纯度98%)、乙二醇单甲醚(纯度99.7%)、乙二醇单乙醚(纯度99.6%)、乙二醇甲醚醋酸酯(纯度99.4%)、乙二醇乙醚醋酸酯(纯度99.9%)、二乙二醇单甲醚(纯度99.9%)、二乙二醇单丁醚(纯度99.5%)、乙二醇单丁醚(纯度99.8%)、二乙二醇二甲醚(纯度99.9%)、二乙二醇二乙醚(纯度99.9%)、三乙二醇二甲醚(纯度99.0%)、N,N-二甲基甲酰胺(纯度99.8%)、N,N-二甲基乙酰胺(纯度99.5%)、N-甲基甲酰胺(纯度99.9%)、N-甲基乙酰胺(纯度99.0%)、N-甲基吡咯烷酮(纯度99.5%)、甲酰胺(纯度99.5%)、1,2,3-三氯丙烷(纯度99%)(德国 Dr.Ehrenstorfer公司)。样品均购于市场。
1.2 标准溶液的制备
标准储备液:准确称取标准物质各0.01 g(精确至0.01 mg),用甲醇为溶剂配成1 000 mg/L储备液,于-18 ℃避光保存。
标准工作液:吸取1 mL标准储备液于10 mL容量瓶中,用甲醇定容至刻度,配成100 mg/L的工作液,再逐级稀释至所需浓度。
1.3 样品前处理
取代表性样品,搅拌均匀后,称取0.2 g(精确至0.000 1 g)试样置于反应器中,加入10 mL甲醇,旋紧盖子,于超声波发生器中提取(30±2) min后,冷却至室温,取有机相离心,再用0.22 μm有机微孔膜过滤,装入进样小瓶(需稀释至适合的倍数),供GC-MS分析用。
1.4 仪器检测条件
毛细管柱:DB-WAX MS柱,60 m×0.25 μm×0.25 mm或相当者;程序升温:起始温度60 ℃保持4 min,以15 ℃/min升温至220 ℃保持10 min,进样口温度:230 ℃;进样量:1 μL。
扫描范围:25~300 amu;流量:0.8 mL/min;离子化方式:EI;离子化电压:70 eV;载气:氦气(≥99.999%);进样模式:分流进样;分流比1∶10;溶剂延迟9.0 min;离子源温度:230 ℃;四极杆温度:150 ℃;传输线温度:280 ℃。
1.5 定性定量分析
分别取1 μL混合标准工作溶液和样品测试溶液,按“1.3”方法前处理,“1.4”方法测试分析。通过比较试样与标样的保留时间及组分的质谱图进行定性分析(见表1)。在0.5~50 mg/L范围内,目标化合物的峰面积与其质量浓度均呈良好的线性关系,相关系数均大于0.999。
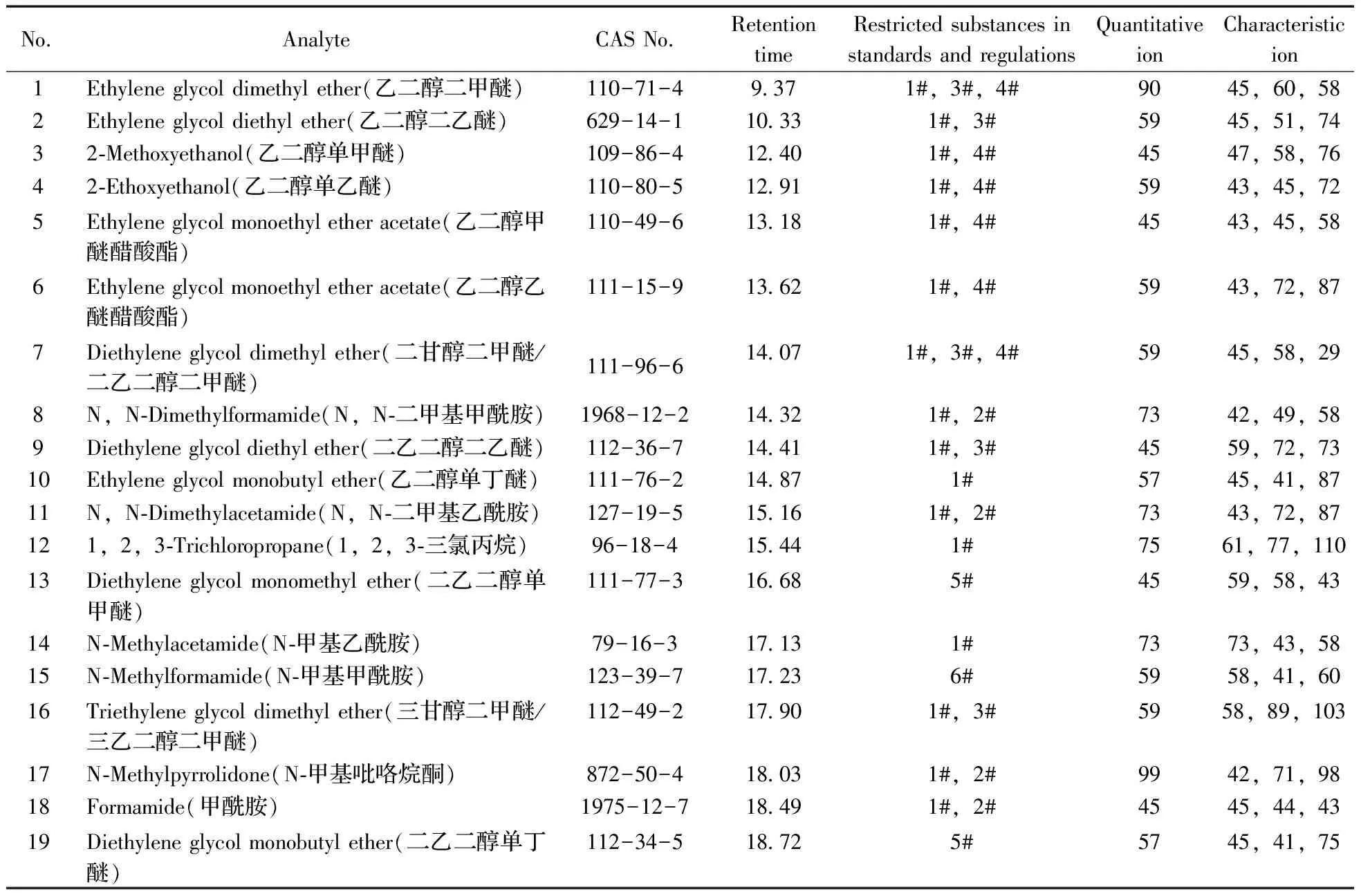
表1 有机溶剂名称及其标准物质的保留时间、限定的法规、定量离子和定性离子Table 1 The names,characteristic ions and quantitative ions of the organic solvent
1# :SVHC;2# :Oeko-Tex Standa100;3#:EPA;4#:ZDHC;5#:REACH regulation;6#:48/2009/EC
2 结果与讨论
2.1 提取溶剂的选择
由于19种分析物的极性不同,所以实验对比了6种代表性的提取溶剂(甲醇、乙腈、丙酮、乙酸乙酯、正己烷、二氯甲烷)的效果。结果显示,选取的6个阳性样品中,甲醇、乙酸乙酯和二氯甲烷的提取效率高于乙腈、丙酮和正己烷,但以乙酸乙酯为提取溶剂时,染整助剂样品大部分呈团聚状,难以提取。以甲醇、二氯甲烷为提取溶剂时,19种分析物的提取效果相差不大,但以二氯甲烷提取时,色谱图上杂质峰较多。因此,本实验选用甲醇作为提取溶剂。
2.2 提取方法的选择
实验对比了振荡提取和超声提取2种提取方法的效果。振荡提取条件:振荡频率150 次/min;超声提取条件如“1.3”所述。分别采用上述两种方法对6个阳性样品进行提取,结果表明,对于多数阳性样品,两种方法无差别,但有些样品超声提取更加符合要求,提取效率更高。因此,本实验选择超声提取。
2.3 提取温度的选择
19种待分析物均属于易挥发性有机溶剂,所以合适的提取温度是保证提取效率的重要条件。采用质量浓度为5 mg/L 的19种有机溶剂混合标准液,比较了20、30、50、70 ℃下的提取效果。结果显示,随着温度的升高,19种分析物的萃取效率均逐渐下降,主要原因可能是高温使分析物挥发。20 ℃和30 ℃时,分析物的回收率较为理想,但30 ℃时的平行性较差。因此,选择最佳提取温度为20 ℃。
2.4 萃取时间的选择
选取6个阳性样品,以甲醇为萃取溶剂,分别超声萃取0、10、20、30、40、50、60 min,比较不同超声时间下的萃取效率。结果表明,随着超声萃取时间的增加,萃取效率明显提高。但萃取时间超过30 min之后,随着萃取时间的继续增加,萃取效率基本保持不变。因此,选择超声萃取时间为30 min。
2.5 色谱柱的选择
19种目标分析物的极性相差较大,所以选择多种不同极性的色谱柱进行比较,即DB-5(弱极性)、DB-17(中等极性)和DB-WAX色谱柱(强极性),在同一进样和分析条件下,对同一浓度的混合标准溶液进行检测。实验结果表明,弱极性色谱柱的响应值低,其分离效果远不及强极性色谱柱,所以实验选用强极性的DB-WAX色谱柱。
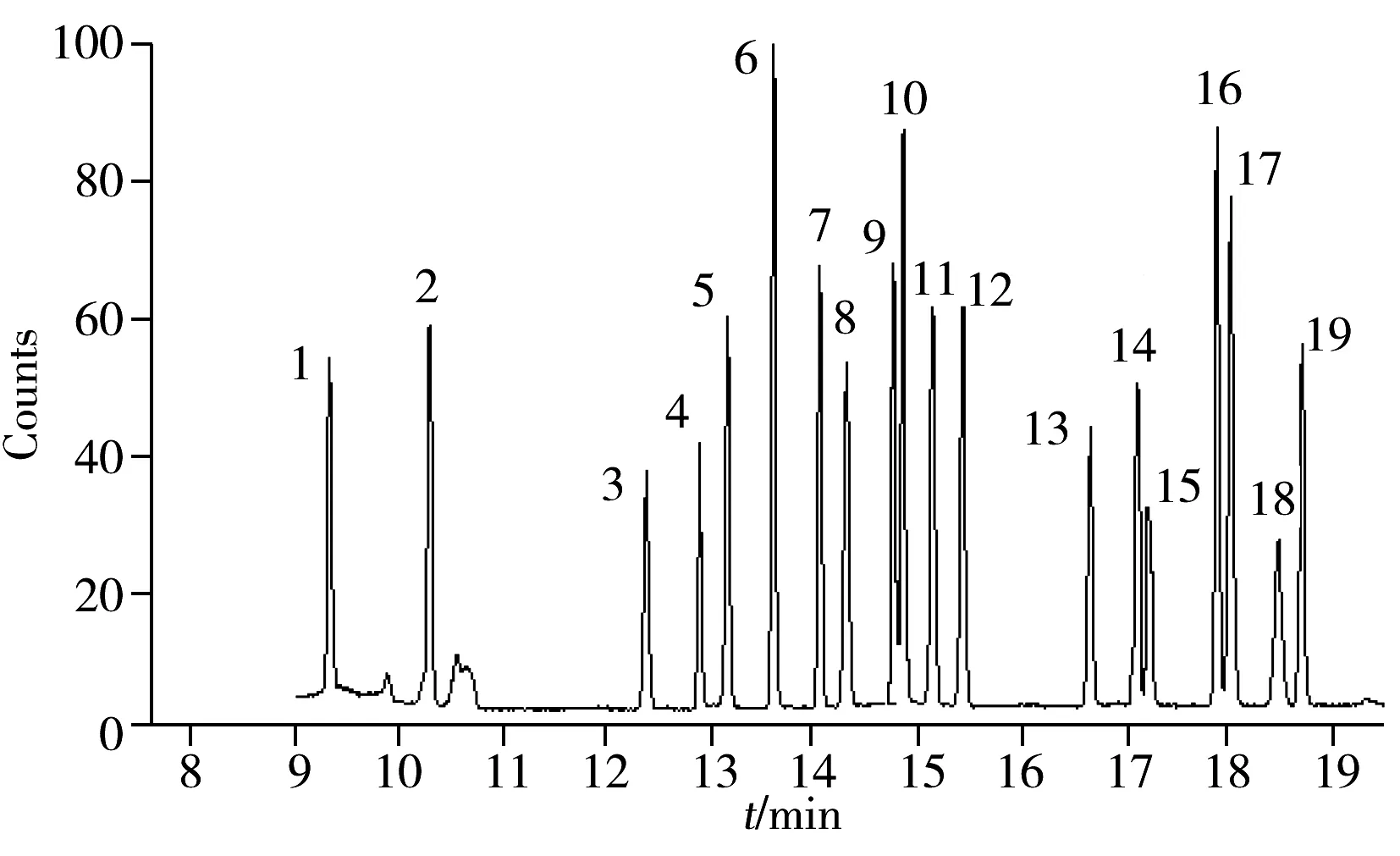
图1 19种有机溶剂混标的总离子流色谱图(含量为5 mg/kg)Fig.1 Total ion chromatogram of 19 organic solvents standard solutionthe numbers noted were the same as those in Table 1
2.6 升温程序的确定
待分析物均为易挥发性有机溶剂,其中有些溶剂的沸点较低,如乙二醇二甲醚的沸点为85 ℃,乙二醇二乙醚的沸点为121.4 ℃,乙二醇单甲醚的沸点为124.5 ℃,乙二醇单乙醚的沸点为135 ℃,所以需确定起始温度以及保持时间。本研究比较了起始温度分别为40、50、60 ℃,以及保持时间分别为0、2、3、4、5 min时19种分析物的出峰情况。结果发现,当起始温度为60 ℃,保持时间分别为4 min或5 min时,19种有机溶剂均能正常出峰,为了节省出峰时间,最终确定起始温度为60 ℃,保持时间为4 min。19种有机溶剂混标的总离子流色谱图见图1。
2.7 方法的线性范围与检出限
配制不同浓度范围的有机溶剂混合标准溶液,经气相色谱分离后,19种有机溶剂均采用外标法定量,在基质匹配的标准溶液中,根据3倍信噪比(S/N=3)计算检出限(LOD)。
采用10次样品空白试验测得的背景响应值的标准差乘以10作为定量下限(LOQ)的估计值,与已知质量浓度的样品进行对照。19种有机溶剂的标准工作点为0.5、1.0、 5.0、10.0、20.0、50.0 mg/L,以峰面积为纵坐标(y),质量浓度(x,mg/L)为横坐标,绘制标准曲线,得到线性方程。结果表明,19种有机溶剂在0.5~50.0 mg/L浓度范围内具有良好的线性(r≥0.999),LOD为0.03~0.10 mg·kg-1,LOQ为 0.10~0.33 mg·kg-1。
2.8 方法的回收率与精密度
采用7#阴性样品,混合标准加标水平分别为0.5、1.0、10.0 mg/kg,按本方法分析检测,每个加标水平平行测试10次。由表2可见,19种有机溶剂的平均回收率为94.3%~101.3%,相对标准偏差(RSD)均小于8.7%,可满足日常分析的需要。
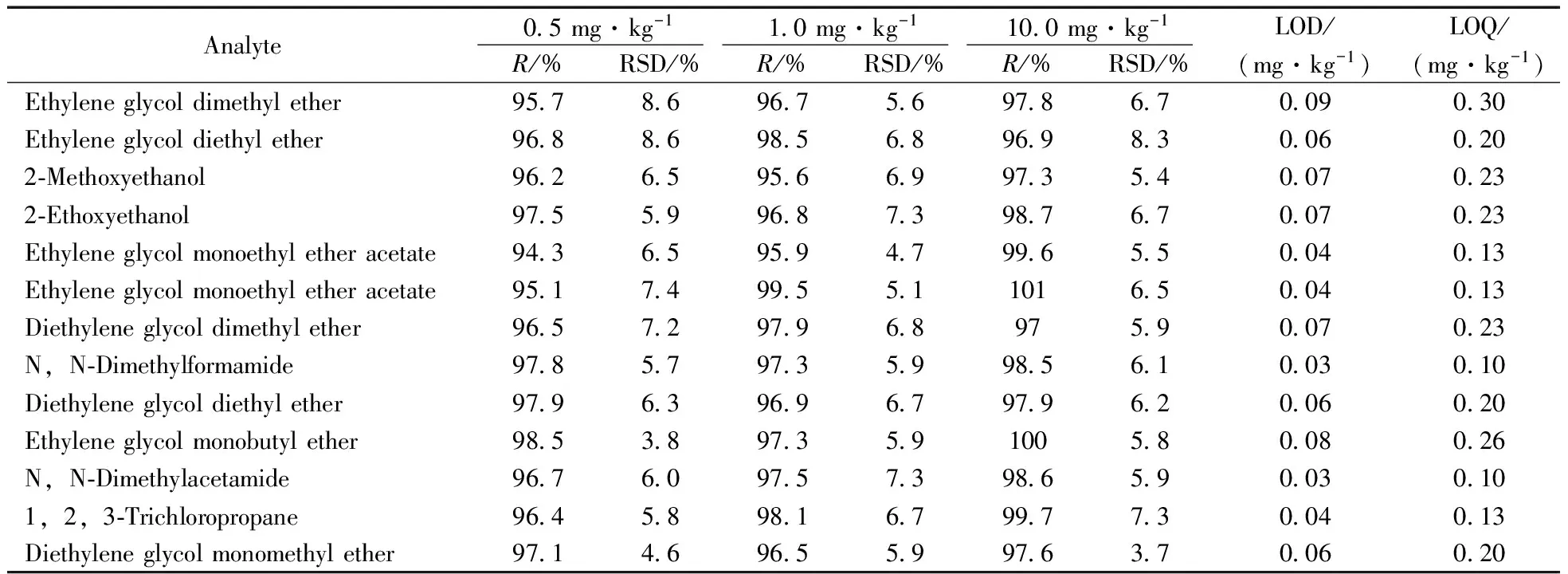
表2 19种有机溶剂的平均回收率、相对标准偏差(n=10)、检出限及定量下限Table 2 Average recoveries,RSDs,LODs and LOQs of 19 organic solvents(n=10)
(续表2)
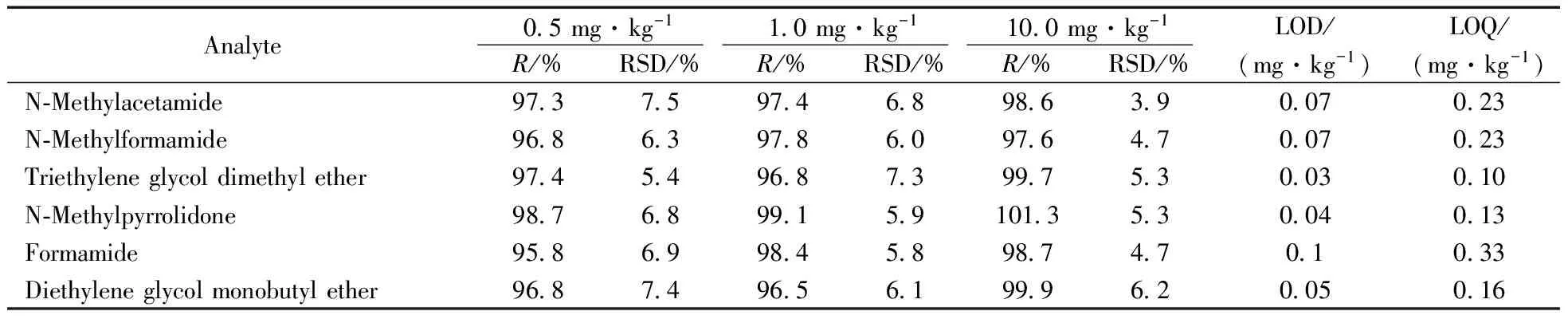
Analyte0 5mg·kg-11 0mg·kg-110 0mg·kg-1R/%RSD/%R/%RSD/%R/%RSD/%LOD/(mg·kg-1)LOQ/(mg·kg-1)N⁃Methylacetamide97 37 597 46 898 63 90 070 23N⁃Methylformamide96 86 397 86 097 64 70 070 23Triethyleneglycoldimethylether97 45 496 87 399 75 30 030 10N⁃Methylpyrrolidone98 76 899 15 9101 35 30 040 13Formamide95 86 998 45 898 74 70 10 33Diethyleneglycolmonobutylether96 87 496 56 199 96 20 050 16
2.9 实际样品检测
应用本方法对现有的染整助剂样品进行测试(表3),其中有5个样品检出乙二醇单丁醚(保留时间为14.88 min),1个样品检出二乙二醇单丁醚(保留时间为18.72 min)。典型阳性样品的色谱图见图2。
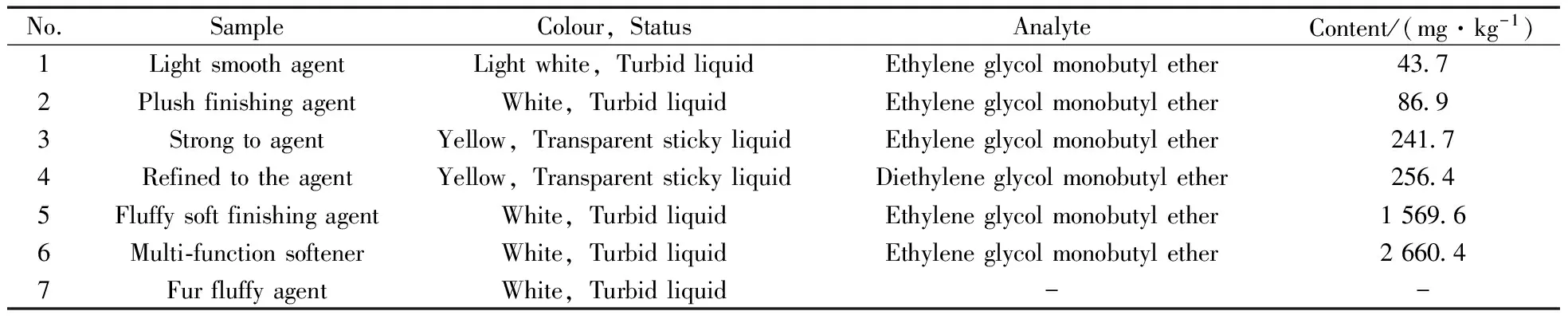
表3 样品编号及描述Table 3 The number and description of the samples
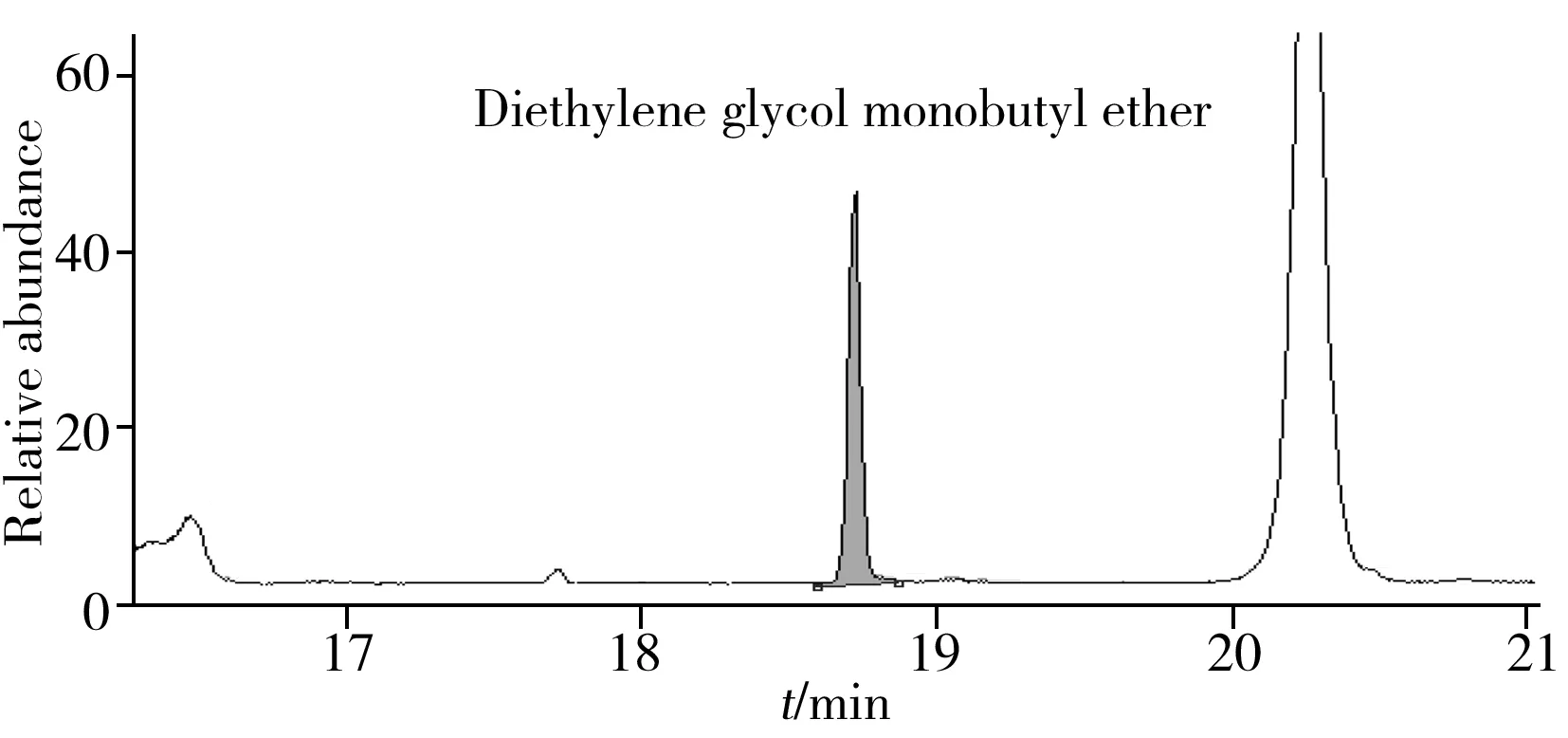
图2 典型样品检出二乙二醇单丁醚的色谱图Fig.2 The chromatogram of a typical positive sample with diethylene glycol monobutyl ether
3 结 论
建立了同时测定染整助剂中19种有害有机溶剂残留量的气相色谱-质谱分析方法,对染整助剂中残留的有害有机溶剂进行了快速筛查和确证。该方法以甲醇作为萃取溶剂,超声萃取,萃取液直接进气相色谱-质谱联用仪测定。该方法的LOQ低至0.10~0.33 mg·kg-1,完全满足检测要求。由于染整助剂广泛应用于日常生活,该方法为相关染料助剂中有机溶剂残留量的检测提供相关参考,也为产品的质量监管提供了重要依据。
[1] Lian Q Y,Liu G.QualityandTechnicalSupervisionResearch(连秋燕,刘贵.质量技术监督研究),2012,24(6):12-16.
[2] Li Z X,Dong W J,Feng J G.GansuSci.Technol.(李仲县,董文江,封聚刚.甘肃科技),2007,23(1):140-142.
[3] Gao D L.StraitPharm.J.(高丹玲.海峡药学),2007,19(7):43-44.
[4] Patricia R,Moiz M,Vijay G.Toxic.Appl.Pharm.,2011,254(2):198-205.
[5] Deconinck E,Canfyn M,Sacrep Y.J.Pharm.Biomed.Anal.,2012,70(1):64-70.
[6] Cheng C,Liu S,Mueller B J.J.Chromatogr.A,2010,1217(41):6413-6421.
[7] Josee R M,Titley M,Bolduc J.J.Chromatogr.B,2004,805(1):77-86.
[8] Chen Z L,Wang J,Chen X Z,Zheng J K.Phys.Test.Chem.Anal.:Chem.Anal.(陈自力,王瑾,陈小珍,郑军科.理化检验-化学分册),2010,46(8):911-916.
[9] Xie P T,Liu M Q.ChinaSurfactantDetergent&Cosmetics(谢佩彤,刘妙青.日用化学工业),2012,42(3):162-166.
[10] He Q S,Liu X L,Wu P,Zhang J C,Cao Y H,Wang G B,Tian X F,Fan J F.Sino-globalEnergy(何庆生,刘献玲,吴平,张建成,曹玉红,王贵宾,田小峰,范景福.中外能源),2013,18:81-84.
[11] Kong X W,Lin Q Q.TextileAuxiliaries(孔祥威,林琼秋.印染助剂),2013,30(5):48-50.
[12] Wang Z,Han C,You F M,Huang H X.FlavourFragranceCosmetics(王征,韩超,游飞明,黄红霞.香料香精化妆品),2013,10(5):24-28.
[13] Che W J,Shen J,Wang Y Q,Wang X D,Pu J.J.Instrum.Anal.(车文军,沈珺,王燕芹,王小丹,浦婕.分析测试学报),2017,36(5):697-700.
[14] Wang C Y,Zhang W Y,Li L X,Shen Y L,Lin J F,Xie T T,Zhu N Q.Chin.J.Chromatogr.(王成云,张伟亚,李丽霞,沈雅蕾,林君峰,谢堂堂,褚乃清.色谱),2014,32(8):890-896.
[15] Tang J,Qi Y,Ding Y C,Cao X Z.J.Chin.MassSpectrom.Soc.(汤娟,齐琰,丁友超,曹锡忠.质谱学报),2015,36(1):59-65.
[16] Du Y Y,Shao Y W,Zhao X,Chen Z Q.Dyeing&Finishing(杜英英,邵玉婉,赵霞,陈志强.印染),2014,(14):42-45.
[17] Ren X X,Li Z W,Wang H,Wang H M,Tian G C.Chem.Res.Appl.(任祥祥,李支薇,王华,王海鸣,田光超.化学研究与应用),2016,28(9):1305-1307.
[18] Zhu H H,Liu J F,Chen Z Z,Shao C J,Yin D W.ChinaLeather(祝惠惠,刘君峰,陈智栋,邵晨杰,尹大伟.中国皮革),2013,42(17):14-16.
[19] Wang C Y,Shi Q Y,Zhu N Q,Lin J F,Xie T T.J.Instrum.Anal.(王成云,施钦元,褚乃清,林君峰,谢堂堂.分析测试学报),2016,35(4):380-387.
猜你喜欢
纺织标准与质量(2022年5期)2022-10-27 06:52:46
纺织标准与质量(2022年3期)2022-08-10 09:11:20
纺织标准与质量(2022年2期)2022-07-12 06:12:56
冰雪运动(2021年2期)2021-08-14 01:54:20
广州化工(2020年5期)2020-04-01 01:24:58
中国盐业(2018年23期)2018-03-30 01:29:28
癌变·畸变·突变(2016年4期)2016-08-22 05:52:34
中国塑料(2015年5期)2015-10-14 00:59:48
食品科学(2013年23期)2013-03-11 18:29:49
上海电机学院学报(2013年3期)2013-03-11 18:08:00
