
PdNi/C低温高效催化湿式氧化无害化处理氨氮废水
2018-02-06 05:57王子丹HameedSohaib张诺伟陈秉辉
厦门大学学报(自然科学版) 2018年1期
王子丹,Hameed Sohaib,张诺伟,陈秉辉
(厦门大学 化学化工学院,醇醚酯化工清洁生产国家工程实验室,福建厦门361005)
高氨氮含量的水体易富营养化而造成水体黑臭,破坏水体生态平衡.含氨氮的生活和工业用水前处理的难度和成本都大幅增加,甚至对人体产生毒害作用.水体的氨氮污染是影响我国地表水环境质量的重要因素之一[1],开发绿色高效的氨氮废水无害化处理技术是废水处理的重要组成部分.催化湿式氧化技术仅利用空气或氧气即能一次性将氨氮催化转化为无害的N2,不需引入其他化学物质即无二次污染;且可以密闭处理废水,避免了开放式废水池向周围散发异味的缺点;同时处理后的废水有可能作为工艺水回收利用,因而其综合性能比目前常用的废水处理方法更符合现代发展的需求.但催化湿式氧化技术也有其缺点,即废水的处理过程往往需要在较高的温度下进行.反应温度的提高将增加能耗,虽然能耗的问题可以通过合理设置处理位置及利用工业过程的废热等手段加以克服,但高反应温度容易产生难以处理的酸.酸的形成不仅给污染物的消除带来困难,还对处理设备提出了更高要求而使处理费用上升.可见,如何在较低的反应温度下实现催化湿式氧化处理氨氮废水,是该技术能更为广泛应用的基础,而其技术的核心在于催化剂.
目前常用的催化剂有负载型Cu催化剂、NiAl2O4、复合金属氧化物和负载型贵金属(Ru、Pt、Pd等)催化剂.负载型Cu催化剂具有较好的氨氮去除性能,但存在活性组分易于流失而失活的问题[2].NiAl2O4在氨氮催化反应中有很好的稳定性与选择性,但氨氮去除率仅为20%[3].复合金属氧化物也是研究较多的一类催化剂,但其性能远低于负载型贵金属催化剂[4].负载型贵金属催化剂因具有优良的催化性能,特别是在低温下具有比一般金属催化剂更好的催化性能而被广泛研究.Qin等[5]的研究结果表明,Pd/Al2O3和Ru/Al2O3具有相近且优于Pt/Al2O3的催化性能;Barbier等[6]则发现Ru/CeO2比Pt/CeO2和Pd/CeO2具有更高的催化活性.此外,Chen 等[7]研究表明与TiO2、Al2O3和MCM-41相比,活性炭(AC)更适合作为氨氮催化湿式氧化催化剂的载体.Pd/AC在相对温和的条件(180 ℃)下能将废水中的氨氮100%去除,即使在150 ℃下也能去除80%以上的氨氮[8].
已有文献[5,9]和本课题组的研究结果表明,氨氮催化湿式反应遵循L-H机理(式(1)~(8),其中*表示活性位点),一般认为O2首先在催化剂表面被活化生成活性氧物种,而后NH3于表面活性氧物种上被活化[5,9];被活化的HNO*可与NH*反应生成N2,或进一步氧化生成副产物NO2-和NO3-,因而表面活性氧物种不仅可活化氨氮,影响氨氮的反应活性,而且决定氨氮的最终氧化产物.

(1)
(2)

(3)
(4)
(5)
(6)
(7)
(8)
具有中等亲氧能力的Ru和Pd具有优良的氨氮脱除性能(氨氮转化率>99%,N2选择性>95%),而亲氧能力低的Pt和亲氧能力高的Mo则使氮氧化合物的选择性高达46%和36%[5].Lousteau等[10]的研究结果表明,催化剂表面的氧覆盖度是氨氮催化转化性能的决定因素,增加氧覆盖度会降低催化活性和N2选择性.一般来说,随着亲氧性能的增强,催化剂表面氧物种的浓度升高而反应活性降低.调变催化剂的亲氧性能可有效提升催化剂的氨氮催化湿式氧化活性.本课题组的前期研究结果表明,Ru和Cu的结合调变了催化剂表面氧物种的特性,从而提高了催化剂的催化性能及稳定性[11].
在贵金属催化剂中,Pd基催化剂具有最好的N2选择性[10].Ni基催化剂也被应用于氨氮的催化湿式氧化,但活性较低[3].基于本课题组的前期研究结果[11]推测,如果将Pd和Ni结合,有望研制出低温高效的氨氮催化湿式氧化催化剂PdNi/C.目前,对PdNi/C催化剂的研究集中在电化学领域.Liu等[12]的研究结果表明PdNi/C对甲醇的电催化氧化性能优于Pd/C催化剂.Holade等[13]研制的Pd60Ni40/C催化剂对甘油的电催化氧化表现出卓越性能.在乙醇燃料电池中,PdNi/C催化剂的活性和稳定性优于Pd/C催化剂[14].PdNi/C催化剂在丙烯醇氧化[15]、甲酸的电催化氧化[16]等方面亦表现出优异的催化氧化性能.但未见有将PdNi/C催化剂应用于氨氮催化湿式氧化的研究.本研究基于贵金属和非贵金属亲氧性能的差异,利用Pd和Ni的协同作用,采用化学还原法制备出低温高效的PdNi/C双金属催化剂,考察了其氨氮催化湿式氧化性能,并对催化剂进行物化性质表征,讨论其构效关系.
1 实验部分
1.1 试 剂
氨水(质量分数为25%)、水合肼(质量分数为85%)、氯化钯、氯化镍、氯化铵和氢氧化钠,以上试剂均为分析纯,购自国药集团化学试剂有限公司.
1.2 催化剂的制备
单金属催化剂的制备:称取0.5 g C于锥形瓶中,加入所需量的水、氨水和1 mL水合肼,搅拌1 h后加入2.6 mL氯化钯(10 mg/mL)或2.6 mL氯化镍(0.1 mol/L)前驱盐溶液,搅拌1 h后过滤、洗涤、烘干制得3%Pd/C或3%Ni/C催化剂(其中3%为Pd或Ni的理论质量分数,实际负载量经原子发射光谱(ICP)测试与理论值相当,下同),使用前在氢气氛围下200 ℃焙烧处理2 h.
双金属催化剂制备:称取0.5 g C于锥形瓶中,加入所需量的水、氨水和1 mL水合肼,搅拌1 h后依次加入1.7 mL氯化钯(10 mg/mL)和0.9 mL氯化镍(0.1 mol/L)前驱盐溶液,搅拌1 h后过滤、洗涤、烘干制得2%Pd1%Ni/C催化剂,使用前在氢气氛围下200 ℃焙烧处理2 h.改变前驱盐溶液的量,采用相同的方法制备1%Pd2%Ni/C和1.5%Pd1.5%Ni/C催化剂.
1.3 催化剂的评价方法
称取0.1 g催化剂于容积为100 mL的快开式微型高压反应釜(安徽科幂机械科技有限公司)中(图1),加入10 mL模拟废水(氨氮的质量浓度为1 000 mg/L,pH=12),开启磁力搅拌,并往反应釜中通入空气(2 MPa),在一定温度下反应3 h后,将反应釜置于冰水中迅速冷却.
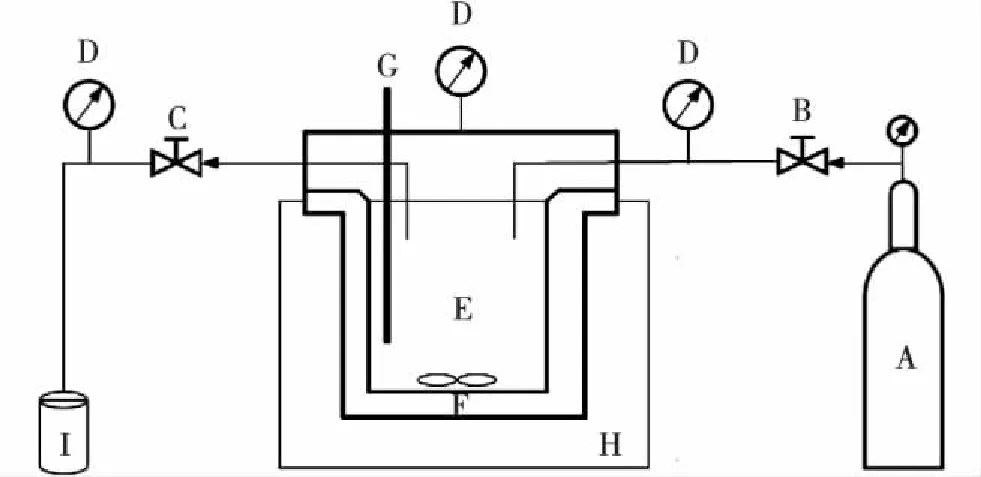
A.空气;B.进气阀;C.排气阀;D.压力表;E.高压反应釜; F.搅拌磁子;G.热电偶;H.加热套;I.尾气吸收液.图1 实验装置简图 Fig.1Schematic diagram of experimental apparatus
利用紫外分光光度计,根据国家标准方法[17]检测反应前后废水中的氨氮含量.反应的主产物为N2,副产物为NO2-和NO3-.反应气相部分用集气袋收集后通过装有Porapack T 和Molsieve 5A填充柱的GC-9160气相色谱仪进行分析[11],除N2外,未检测到NO、N2O等氮氧化物副产物.NO2-和NO3-通过高效液相色谱检测分析,采用美国DIONEX C18色谱柱,流动相为磷酸氢二铵-磷酸缓冲液(pH≈3)和乙腈(体积比90∶10),流速1 mL/min,柱温35 ℃,波长210 nm.氨氮转化率和N2选择性等按下列公式计算:
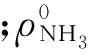
1.4 催化剂的物化性能表征
样品的物相结构分析在Rigaku Ultima Ⅳ X射线衍射(XRD)仪上完成,Cu靶,Kα辐射,工作电压35 kV,工作电流20 mA,扫描速度10 (°)/min,扫描范围10°~90°;X射线光电子能谱(XPS)测试在Quantum 2000 X射线扫描微探针电子能谱仪上进行,以单色化Al Kα为X射线源;利用CO程序升温还原(CO-TPR)分析催化剂的氧化还原特性.
2 结果与讨论
基于本课题组的实验条件优化结果[11],首先在反应温度为140 ℃,反应压强(空气)为2 MPa,反应时间为3 h的条件下考察了3%Ni/C、3%Pd/C和2%Pd1%Ni/C催化剂的氨氮催化湿式氧化性能,结果见表1.可以看出,3种催化剂均具有良好的N2选择性(>90%),但反应活性相差很大.3%Ni/C、3%Pd/C和2%Pd1%Ni/C催化剂的氨氮转化率分别为50.6%,86.4%和99.0%.与3%Ni/C相比,3%Pd/C催化剂具有更高的反应活性,而Ni和Pd的结合则能有效提升催化剂的催化氧化性能.目前,未见文献报道有催化剂能在140 ℃达到如此高的氨氮转化率.可见,Pd和Ni的协同作用使得双金属催化剂在较低贵金属用量的前提下,得到较高的氨氮转化率.在保持金属总负载量为3%的前提下,对Pd和Ni的质量比进行优化,结果如表2所示.可以看出,氨氮转化率随着Pd负载量的增大而增大,当Pd和Ni的质量比为2∶1时,催化剂催化氧化氨氮反应的活性最优,氨氮转化率接近100%.
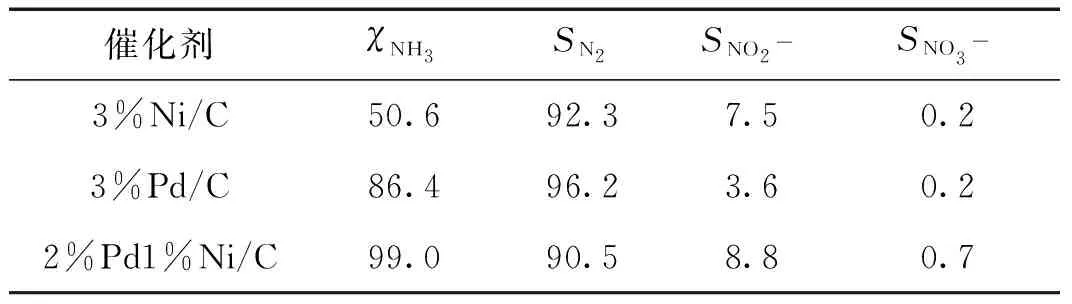
表1 Pd和Ni协同作用对氨氮催化湿式氧化性能的影响Tab.1 Effect of synergy between Pd and Ni on the catalytic performance for catalytic wet air oxidation of ammonia %
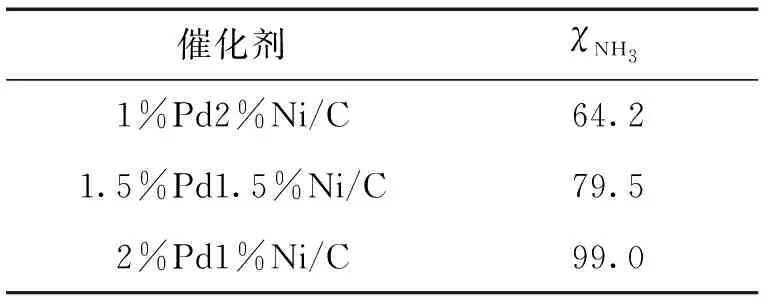
表2 Pd和Ni比例对催化氧化氨氮反应活性的影响Tab.2 Effect of Pd and Ni proportions on catalytic oxidation of ammonia %
在不同反应温度条件下进一步考察了Ni的添加对催化性能的改善作用,结果如图2所示.温度对反应活性有较大影响,随着反应温度升高,2%Pd1%Ni/C和3%Pd/C催化剂的氨氮转化率都逐步增加,但与3%Pd/C相比,2%Pd1%Ni/C催化剂在各个反应温度都具有更优的催化活性:120 ℃时,2%Pd1%Ni/C催化剂的氨氮转化率为81.0%,而3%Pd/C催化剂的氨氮转化率仅为68.0%;当温度升至130 ℃时,二者分别升至90.7%和77.9%;140 ℃时,2%Pd1%Ni/C催化剂的氨氮转化率接近100%,而3%Pd/C催化剂则仅为86.4%.Ni的添加使得双金属催化剂能在较低的反应温度下得到较高的催化活性.
图3为3%Pd/C、3%Ni/C和2%Pd1%Ni/C催化剂的XRD谱图.可以看出:3%Ni/C催化剂未出现任何Ni物种的衍射峰,表明催化剂上的Ni物种高度分散;3%Pd/C催化剂上检测到金属Pd,且衍射峰尖锐,表明催化剂活性组分以Pd形态存在,且金属Pd明显团聚;2%Pd1%Ni/C催化剂上也检测到了金属Pd,但与3%Pd/C相比,其Pd的衍射峰强度明显减弱,表明Ni的添加能有效地促进Pd的分散.较好的分散性可能是2%Pd1%Ni/C双金属催化剂比3%Pd/C单金属催化剂具有更好催化性能的原因之一.
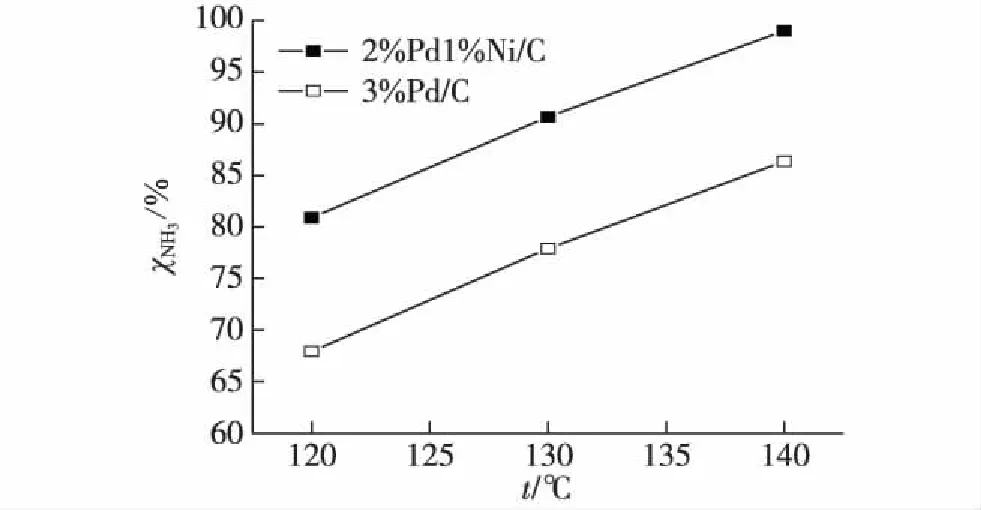
图2 反应温度对2%Pd1%Ni/C和 3%Pd/C催化活性的影响 Fig.2Effect of reaction temperature on catalytic activity of 2%Pd1%Ni/C and 3%Pd/C
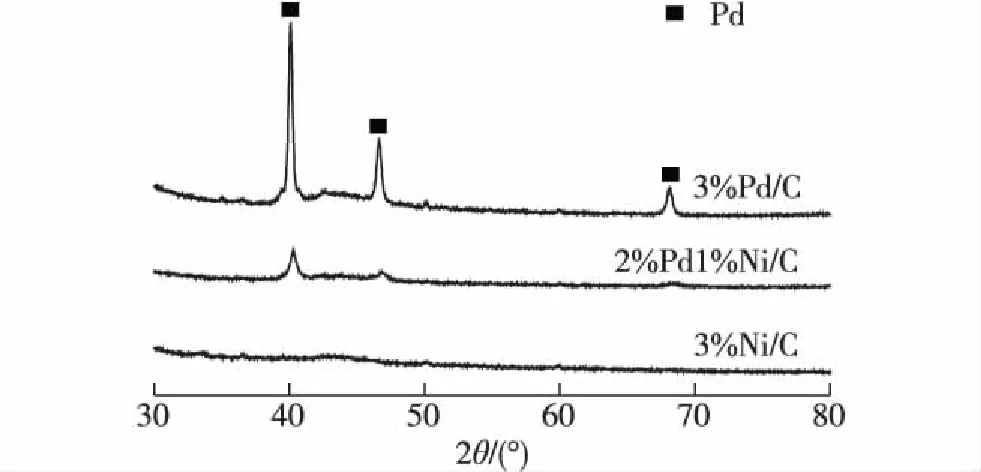
图3 3%Pd/C、2%Pd1%Ni/C和3%Ni/C的XRD谱图 Fig.3XRD patterns of 3%Pd/C, 2%Pd1%Ni/C and 3%Ni/C
利用CO-TPR技术考察了3%Pd/C、3%Ni/C和2%Pd1%Ni/C催化剂的氧化还原特性.实验前催化剂在氧气气氛、140 ℃下预处理30 min.在CO-TPR过程中CO2除了来自氧物种的还原(如CO+PdO→Pd+CO2),还可能来自水煤气变换反应(CO+H2O→CO2+H2).考虑到只有水煤气变换反应产生H2,故可根据H2的信号判断CO2的来源[18].
图4(a)为CO-TPR过程中H2信号随温度的变化曲线.可以看到:在考察的温度范围内,3%Pd/C和2%Pd1%Ni/C催化剂没有出现明显的H2生成峰,表明在上述温度范围的CO-TPR过程中基本没有发生水煤气变换反应;而3%Ni/C催化剂在(400~600 ℃)出现了H2生成峰,可能的原因是催化剂表面的Ni(OH)2和NiOOH还原生成了H2O,从而导致了水煤气变换反应的发生.
图4(b)为CO-TPR过程中CO2信号的检测结果,与图4(a)比较可知,3种催化剂的CO2信号主要来自于催化剂表面氧物种的还原.3%Pd/C催化剂CO2生成峰的温度最低(330 ℃附近),但峰面积最小,表明该催化剂表面氧物种的反应活性最高,但氧物种浓度最低;3%Ni/C催化剂CO2生成峰的温度最高,最强峰出现在500 ℃附近,且峰面积最大,表明该催化剂表面氧物种浓度最高,但反应活性最低;而2%Pd1%Ni/C双金属催化剂的CO2生成峰的温度和峰面积都介于两者之间,表明双金属催化剂表面氧物种具有适中的反应活性和浓度,同时也表明Pd和Ni之间存在相互作用.氧物种的特性(反应活性和浓度)与催化性能息息相关:3%Pd/C催化剂表面氧物种具有最高的反应活性,但浓度低,因而与2%Pd1%Ni/C双金属催化剂相比,催化相对活性较低;尽管3%Ni/C催化剂表面氧物种浓度最高,但反应活性低,不足以活化氨氮,因而其催化性能最低;而2%Pd1%Ni/C双金属催化剂则因表面氧物种的反应活性和浓度适中而具有高的催化活性.
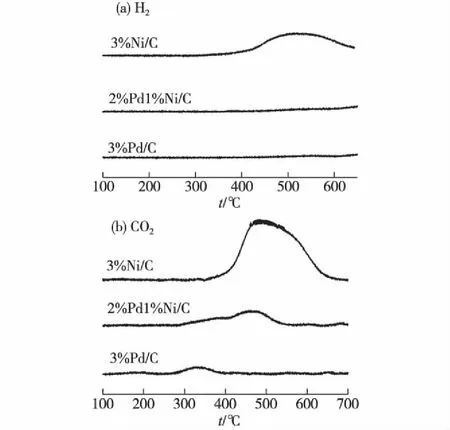
图4 3%Ni/C、2%Pd1%Ni/C和3%Pd/C 催化剂的CO-TPR结果 Fig.4CO-TPR profiles of 3%Ni/C,2%Pd1%Ni/C and 3%Pd/C catalysts
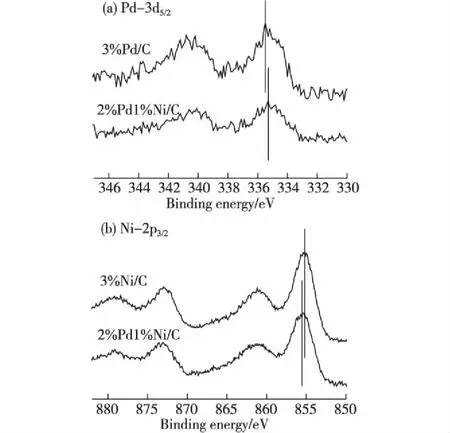
图5 3%Pd/C、3%Ni/C和2%Pd1% Ni/C的XPS表征结果 Fig.5XPS characterization results of 3%Pd/C, 3%Ni/C and 2%Pd1%Ni/C
已有研究表明氨氮催化湿式氧化反应遵循L-H机理[5,9,11,19-20],据此推测,本研究涉及的催化体系也应遵循L-H机理.与气相多相催化体系相比,受水的影响,氨氮催化湿式氧化催化剂的表面活性位以及中间活性物种的表征较难实现,下一步将会继续开展关于表面活性位和中间活性物种的表征工作,以深入研究氨氮催化湿式氧化的反应机理.
3 结 论
与单金属3%Pd/C和3%Ni/C催化剂相比,2%Pd1%Ni/C双金属催化剂具有更好的氨氮催化湿式氧化反应性能,在140 ℃下废水中氨氮的转化率接近100%,即使在120 ℃下也可脱除80%以上的氨氮.这是由于Ni的添加提高了Pd的分散性,并与Pd相互作用,调变了表面氧物种的特性(适中的反应活性和浓度),进而提高了催化剂的氨氮催化湿式氧化性能.尽管制备的2%Pd1%Ni/C催化剂具有良好的催化活性,但稳定性并不十分理想,连续反应3次,氨氮的转化率分别为99.0%,79.5%和42.4%,导致催化剂失活的原因有待进一步深入研究.
催化湿式氧化法具有高效且反应器体积小的优点,适合处理较高浓度的氨氮废水.而生物脱氮方法效率低且需要大面积处理池,适合处理低浓度氨氮废水,其优点是可在常温下处理,处理后的废水可达到很低的氨氮浓度.因此,二者具有一定的互补性,实际应用中可采取二者相结合的方法:将较高浓度的氨氮废水先在密闭反应器中采用催化湿式氧化法处理,再采用生物脱氮法处理,这样既可防止异味散发到空气中,又能极大地缩小处理池的体积,保证废水达标.
[1] 吴悦颖,文宇立,刘伟江.“十二五”氨氮总量怎么减? [J].环境经济,2013(3):39-41.
[2] 付迎春,钱仁渊,金鸣林.催化湿式氧化法处理氨氮废水的研究 [J].煤炭转化,2004(2):72-75.
[3] KAEWPUANG-NGAM S,INAZU K,KOBAYASHI T,et al.Selective wet-air oxidation of diluted aqueous ammonia solutions over supported Ni catalysts [J].Water Research,2004,38(3):778-782.
[4] IMAMURA S,DOI A,ISHIDA S.Wet oxidation of ammonia catalyzed by cerium-based composite oxides [J].Industrial & Engineering Chemistry Product Research and Development,1985,24(1):75-80.
[5] QIN J Y,AIKA K.Catalytic wet air oxidation of ammonia over alumina supported metals [J].Applied Catalysis B:Environmental,1998,16(3):261-268.
[6] BARBIER J,JR,OLIVIERO L,RENARD B,et al.Catalytic wet air oxidation of ammonia over M/CeO2ca-talysts in the treatment of nitrogen-containing pollutants [J].Catalysis Today,2002,75(1/2/3/4):29-34.
[7] CAO S L,CHEN G H,HU X J,et al.Catalytic wet air oxidation of wastewater containing ammonia and phenol over activated carbon supported Pt catalysts [J].Catalysis Today,2003,88(1/2):37-47.
[8] TAKAYAMA H,QIN J Y,INAZU K,et al.Hydrogen-treated active carbon supported palladium catalysts for wet air oxidation of ammonia [J].Chemistry Letters,1999,28(5):377-378.
[9] LEE D K,CHO J S,YOON W L.Catalytic wet oxidation of ammonia:why is N2formed preferentially against NO3-? [J].Chemosphere,2005,61(4):573-578.
[10] LOUSTEAU C,BESSON M,DESCORME C.Catalytic wet air oxidation of ammonia over supported noble metals [J].Catalysis Today,2015,241:80-85.
[11] FU J L,YANG K X,MA C J,et al.Bimetallic Ru-Cu as a highly active,selective and stable catalyst for catalytic wet oxidation of aqueous ammonia to nitrogen [J].Applied Catalysis B:Environmental,2016,184:216-222.
[12] LIU Z,ZHANG X,HONG L.Physical and electrochemical characterizations of nanostructured Pd/C and PdNi/C catalysts for methanol oxidation [J].Electrochemistry Communications,2009,11(4):925-928.
[13] HOLADE Y,MORAIS C,ARRII-CLACENS S,et al.New preparation of PdNi/C and PdAg/C nanocatalysts for glycerol electrooxidation in alkaline medium[J].Electrocatalysis,2013,4(3):167-178.
[14] SHEN S Y,ZHAO T S,XU J B,et al.Synthesis of PdNi catalysts for the oxidation of ethanol in alkaline direct ethanol fuel cells [J].Journal of Power Sources,2010,195(4):1001-1006.
[15] JIN C C,SUN X J,CHEN Z D,et al.Electrocatalytic activity of PdNi/C catalysts for allyl alcohol oxidation in alkaline solution [J].Materials Chemistry and Physics,2012,135(2/3):433-437.
[16] 高艳伟,邓超,邬冰,等.PdNi/C催化剂的制备及对甲酸的电催化氧化 [J].化学工程师,2010,24(5):1-3.
[17] 沈阳市环境监测中心站.水质 氨氮的测定 纳氏试剂分光光度法:HJ 535—2009 [S].北京:中国环境科学出版社,2009.
[18] ZHU H Q,QIN Z F,SHAN W J,et al.Pd/CeO2-TiO2catalyst for CO oxidation at low temperature:a TPR study with H2and CO as reducing agents [J].Journal of Catalysis,2004,225(2):267-277.
[19] LEE D K.Mechanism and kinetics of the catalytic oxidation of aqueous ammonia to molecular nitrogen [J].Environmental Science and Technology,2003,37(24):5745-5749.
[20] WANG Z,HAMEED S,WEN Y,et al.The effect of weak acid anions on the selective catalytic wet air oxidation of aqueous ammonia to nitrogen [J].Scientific Reports,2017,7(1):3911.
猜你喜欢
化工管理(2022年13期)2022-12-02
机械工业标准化与质量(2022年6期)2022-08-12
环境卫生工程(2021年4期)2021-10-13
航天工业管理(2020年9期)2020-12-28
重型机械(2020年2期)2020-07-24
建材发展导向(2019年11期)2019-08-24
中国调味品(2017年2期)2017-03-20
中国资源综合利用(2016年11期)2016-01-22
中国石油大学学报(自然科学版)(2015年2期)2015-11-10
中学化学(2015年2期)2015-06-05
