
基于位置信息的非比对序列聚类方法
2018-01-03 01:57:53徐彭娜江育娥
计算机应用与软件 2017年12期
魏 静 徐彭娜 江育娥 林 劼
(福建师范大学软件学院 福建 福州 350108)
基于位置信息的非比对序列聚类方法
魏 静 徐彭娜 江育娥 林 劼*
(福建师范大学软件学院 福建 福州 350108)
非比对序列相似性模型直接采用序列自身的统计信息来计算序列之间的相似度,具有运算速度快、聚类结果准确等优点。提出一种基于位置信息的非比对序列相似性模型,通过提取K词模型中每个词的Local Frequency(LF),计算对应K词的LF熵,并结合K词频率进行序列的特征提取,应用于蛋白质聚类。实验结果表明该方法能够有效地提取序列的信息,提高聚类的准确率。
K-词 LF熵 K-means聚类 位置信息
0 引 言
在生物信息研究领域中,序列的特征提取和相似性检测是十分重要的,对大量的生物序列进行聚类分析是生物数据挖掘的研究方向之一,其应用前景十分广泛。近年来,快速发展的生物科学技术使得生物信息处理领域的数据资源快速增长。传统的比对序列方法面对庞大的生物数据集,越来越不能满足需求,而且比对序列法的计算过程是非常耗时的,而非比对法直接利用序列本身所携带的信息来计算序列之间的相似度。因此,在序列相似性研究中,非比对方法越来越受欢迎。
序列比对方法一般是采用BLAST[1]和FASTA[2]这两类算法,他们均是基于识别“比对种子”的启发式算法,并得到广泛的应用。非比对法包含有图形表示法、概率统计方法、K-词的方法、文本压缩方法以及信息理论方法等。Dai等[3]在序列比较的非比对法研究中,通过集成生物序列中字的重叠结构和背景信息,提出了一种新的统计方法。Bonham-Carter等[4]等在文献中解释几个不同领域的非比对分析方法,并提供了几个明确的例子来演示应用程序。Randic等[5]等通过距离矩阵法把蛋白质序列用图形方法表示。在许多非比对方法研究中均是基于K-词频率,K-词算法是非常受欢迎的非比对法。一条生物序列通过一个长度为K的滑动窗口,在窗口中的这一段序列是一个K-词。K-词频率算法是计算每一个词出现的频数,建立基于频率值的特征向量。基于K-词频率的非比对方法通常是把序列中的每个K-词看作为独立的个体,从而忽略词与词之间的联系和整体性。序列特征不仅仅是指词频特征,K-词出现的位置间的相关性和碱基的性质都包含大量的序列信息。余宏杰[6]根据序列的顺序属性,对K-词定义一种复合变换,提出一种新的非比对序列的比较方法。邓伟[7]在研究序列相似性和构建进化树时,结合核苷酸的化学结构分类,将已有的混沌游走表达模型进行了改进,并提取DNA序列特征构建特征向量。Leimeister等[8]在非比对序列比较法研究中,提出使用间隔词来降低相邻词匹配间的统计相关性,通过模式的“匹配”和“不相关”位置定义,使用递归散列和位操作快速实现。
本文通过词的位置信息计算LF值并计算熵值,得出每条序列对应词的LFE(即Local Frequency Entropy的缩写),结合蛋白质序列K-词的词频特征构建序列的特征向量。实验结果证明基于位置信息的非比对序列聚类方法是一种非常有效的序列聚类方法。
1 相关工作研究
众所周知,一般的基于K-词频率的非比对方法研究只包含词频信息,并不能够完全地提取出序列的特征,而特征向量的不完整性则会严重影响聚类的效果。生物序列中所包含的信息不仅仅是序列的组成,每个K-词的位置分布以及每个词产生的顺序都蕴含了大量的信息。
Bao等[9]根据Local Frequency计算熵值,根据碱基的分类,位置分布特征和发生频率,将一条DNA序列划分为三个新序列,测量12维空间中的相似性,称为Category-Position-Frequency 模型(简称CPF模型)。Li等[10]根据词频在整个原始序列中的位置信息,首次提出Global Frequency(简称GF),计算序列的相似性。许景皓[11]在进行蛋白质结构功能预测时,利用氨基酸的位置信息计算氨基酸距离对,检测蛋白质远程同源性和计算蛋白质序列相似性。杨希武[12]通过获取序列中K-词的位置间的相互关系,简化得到K-词的位置序列并按照数值大小进行排序,序列间的相似性通过欧氏距离进行计算。吴迪[13]在对序列进行聚类操作前,将序列预处理为等长的向量,依据序列元素的相似度和Top-k最大序列模式提出一种新的序列聚类算法。Shi等[14]提出的TSM方法,利用核苷酸碱基及其化学性质将一条DNA序列分为三条符号序列,提取DNA序列的特征并进行序列相似性分析。黄玉娟[15]在进行DNA序列研究时,根据 K-词频度和其位置信息,定义一个新的概率分布,为了降低碱基突变造成的影响,新的概率分布减去了背景概率,它们之间的相对差异性可以用来表征一条DNA序列。胡清铭[16]根据氨基酸残基的位置,提出一种新的氨基酸残基位置特征提取方法,获得的特征向量维数低、计算简单,而且能够有效地提取氨基酸的位置信息。Kurgan等[17]在利用氨基酸组成方法的基础上,根据氨基酸的位置信息,进行预测蛋白质结构和功能。唐杰[18]在序列相似性的比较上提出基于K-词位置的序列非比对方法,用机器学习的一些方法来处理数据,然后对序列进行分类。
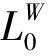
(1)
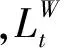
基于位置信息的非比对序列聚类方法,综合考虑每个K-词的位置信息和词频特征,尽可能完整地从生物序列中提取有效的特征,能够准确地反映出生物序列信息和功能之间的关联性。
2 基于位置信息的非比对序列的方法
本文的方法首先是将序列采用K-词模型进行映射,预处理字长为K,从而获得|Σ|k个特征词,即K-词,Σ是序列符号集合,|Σ|是序列符号集合大小。统计每个K-词出现的位置及其频数,通过式(2)计算出每个词的LF值,LF值即为K-词出现的相邻两个位置之间差的倒数;依次对所有词根据式(2)进行该词的LF值计算,并得到每条序列的每个词的LF值序列,即每条序列经过计算得到|Σ|k个LF值的数值序列。
定义LF值V为:
(2)
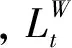
根据得到的LF值,由式(3)、式(4)、式(5)、式(6)计算出LF熵,即LFE。对于词W的LFE计算过程如下:
1) 序列的部分和,令:
(3)
即:
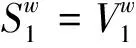
2) 计算序列S的总和Z,即:
(4)
3) 计算每个位置t的离散概率,即:
(5)
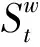
4) 计算该序列的熵LFE的值VE,即:
(6)
结合LFE和词频信息作为序列的特征,来提取序列的信息。通过计算N条序列的VE构成N×|Σ|k的特征矩阵,结合蛋白质序列的每个K-词的词频特征,构成N×2×|Σ|k的特征矩阵。通过K-means聚类方法对序列特征进行聚类。根据聚类结果计算出准确率和召回率。通过F-measure评价标准对模型进行评价。
LFE强调每个词出现的位置顺序和这个词在序列中的局部密度,一个LF不能包含这个词的全局信息,但是一系列的LFE可以更加精确和清晰地表示出这个K-词的全局分布。采用LF综合考虑到在转换后的特征空间包含足够的原始序列信息,避免序列信息的丢失。以Local Frequency为基础的计算而得的熵值能精确地反映序列的结构信息。序列的相似性作为生物信息学中的基本度量,在许多场合中都会有应用,包括预测一段未知序列的作用和功能、构建生物或者物种的系统进化树、分析物种的同源性等。
3 实验与结果分析
3.1 数据来源
实验数据来源于PBIL的HOMOLENS[21]数据库中的蛋白质数据集,从中随机选取20个family进行实验,同时为了验证本实验方法随着实验数据集的增加依然有效,随机挑选出100个、200个、300个family进行同样的实验操作。虽然每个family中的蛋白质序列数量和序列的长度大小都有所区别,但是同一个family中的蛋白质序列都具有一定的相似性。
3.2 实验步骤
在进行实验之前对数据进行整理,将某些序列中含有的字母X删除,因为字母X代表任意的氨基酸,无法对其进行相关的统计与处理。
将原始蛋白质序列按着K-词模型进行映射,由于蛋白质序列是由20个字母组成,按照字母表顺序依次映射为数值0到19,为了实验的方便我们取K-词模型的K值为1,从而获得20k个待处理字,即K-词。
(1) 将从原始数据集中获取的蛋白质序列按K-词模型进行映射得到对应的数字序列,预处理字长为K,获得20k个待处理字。
(2) 统计每条序列中每个K-词的频度和位置,LF的值V等于相邻两个位置相减的倒数。
(3) 根据式(2)计算出所有词对应的LF的值V,再分别根据式(3)、式(4)、式(5)计算出对应的累积和S、总和Z及离散概率P,最后根据式(6)计算出VE。由于V为相邻位置差的倒数,当词在序列中出现的频度小于或者等于2时计算出的VE都为0,无法明确区分出词的各个频度特征,因此在本实验中对于词频小于或等于2的情况进行了单独处理,即频度为0与频度为4,频度为1与频度为3的情况进行对称处理,频度为2时,VE等于0。
频度为1时,
VE=2×(1/2)×log2(1/2)=-1
频度为0时,
VE=3×(1/3)×log2(1/3)=-1.584 963
(4) 计算的VE构成一个N×20k的特征矩阵,结合每个词的频度特征构成一个N×2×20k的特征矩阵。由于蛋白质序列存在长度差异,为了消除序列长度对词频的影响,在本实验中对每个词的词频进行标准化处理。
(5) 对由LFE和词频构成的特征进行K-means聚类。初始聚类中心对K-means聚类算法的聚类结果影响很大,由于每一次随机选取的聚类中心不同,得到的聚类结果也是不同的,为了消除偶然性结果的干扰,进行多次重复实验,取平均值。
3.3 模型评价方法
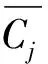
fm是选取的family数。
经过K-means聚类后,通常一个family会被分为几个不同的集群中,在具有相同的family标签的集群的基础上,计算一个family的精确度和召回率。
第i个family的精确度计算公式:
(7)
第i个family的召回率计算公式:
(8)
则第i个family的F值计算公式为:
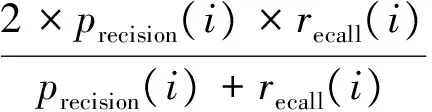
(9)
实验的聚类结果的F值:
(10)
由于K-means聚类结果与所选取的初始聚类中心有很大的关系,为了消除偶然性结果的干扰,选取的family数从5到20时,对每一次随机选定的数据集,重复试验10次,计算出这10次的精确度和召回率以及F值,然后分别计算它们的平均值。
随机分别选取100、200、300 family进行同样的实验,分别计算出F值,精确度和召回率,与Bao等的实验结果进行比较。
3.4 实验结果及分析
在20个family实验过程中使用LFE和词频特征来提取蛋白质序列的特征。蛋白质序列由20个family组成,实验过程中随机抽取5到20个family进行聚类分析,计算F值,并且与Bao等在文献[9]中的CPF模型计算的F值进行对比,另外对这两组数据进行T检验。如图1所示。
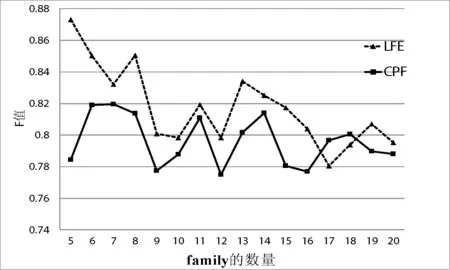
图1 family数从5到20的F值情况
图1是选择的family数从5到20情况下F值的情况。每次选定family数后重复10次随机实验,计算10次F值,取平均值。从图中可以看出在family数量的从5到20的实验过程中,LFE模型所得到的F值要大于Bao等[9]的CPF模型计算的F值,虽然在family数为17和18时效果不理想,但是总体而言本文的LFE模型计算的F值相对较高。
两组F值T检验后,p-value=0.007 662;p值小于0.05,可以确定LFE的聚类效果显著优于CPF模型。
图2和图3分别是选择的family数从5到20情况下精确度和召回率的情况。从图中可以看出LFE的模型计算的精确度和召回率要明显高于CPF模型。
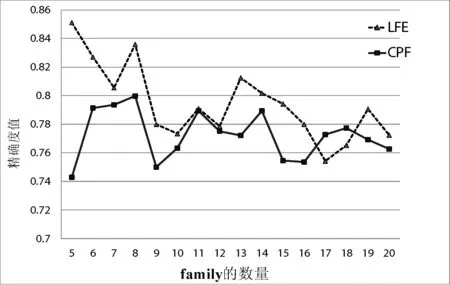
图2 family数从5到20的精确度情况
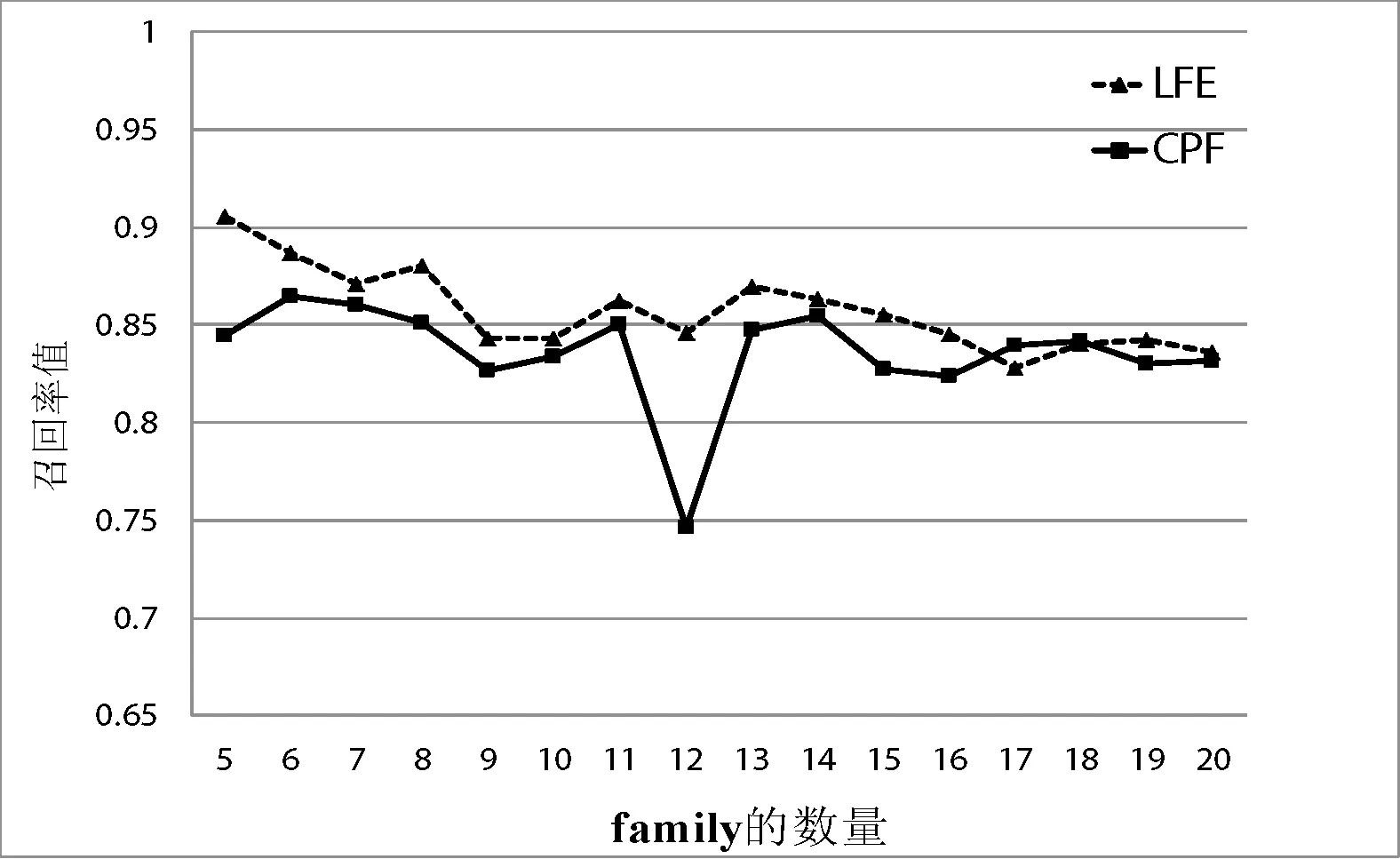
图3 family数从5到20的召回率情况
图4是family数为100、200、300时,重复10次实验的F值、准确率和召回率的平均值,从左到右分别是HOG100、HOG200、HOG300时,LFE方法计算的F值、CPF方法计算的F值,LFE方法计算的准确率、CPF方法计算的准确率,LFE方法计算的召回率、CPF方法计算的召回率。
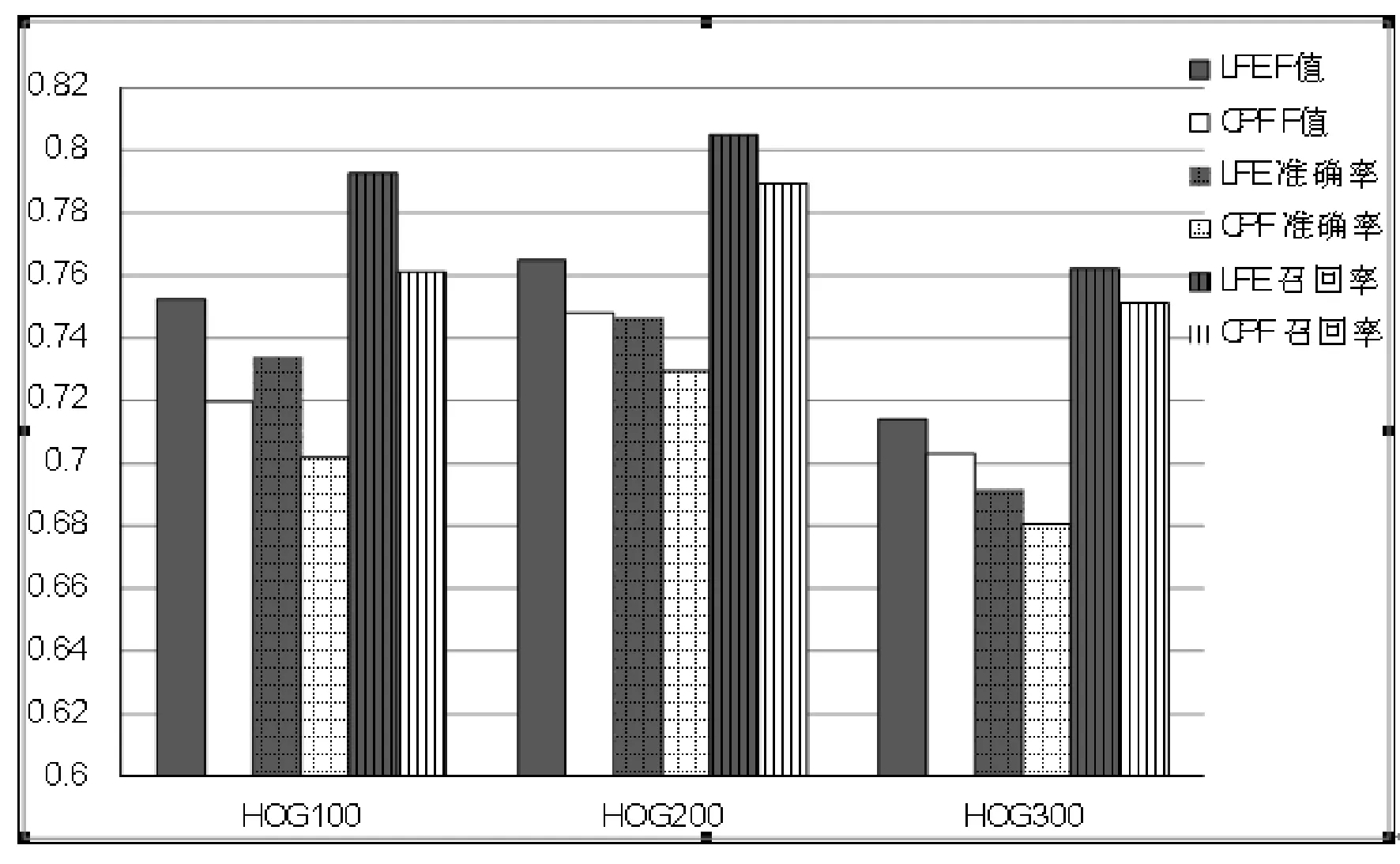
图4 family数为100、200和300时F值准确率和召回率情况
从上图的实验结果可以看出,随着数据集的增加,LFE模型聚类结果的F值、精确度和召回率均比CPF模型好。对HOG100数据集、HOG200数据集、HOG300数据集使用LFE方法、CPF方法得到的F值,对其T检验的p-value值分别为8.17E-05、0.001 496和0.016 31。p值明显小于0.05,可以确定LFE的聚类效果显著优于CPF模型。
4 结 语
本文对蛋白质数据采用了基于位置信息的非比对序列的方法进行聚类。通过K-词模型进行映射得到20k个K-词,将词的位置信息与词频特征相结合作为序列的特征向量,对特征向量进行K-means聚类,计算精确率和召回率,用F-measure对本文的方法进行评估。随机分别抽取20、100、200、300个family的蛋白质序列进行聚类研究,实验结果表明随着实验数据集的增加,本文实验方法同样取得很好的效果,相比于Bao等[9]的CPF模型的聚类结果,本实验的聚类效果显著提高。
[1] Altschul S F,Gish W,Miller W,et al.Basic local alignment search tool[J].Journal of Molecular Biology,1990,215(3):403-410.
[2] Pearson W R.Rapid and sensitive sequence comparison with FASTP and FASTA[J].Method in Enzymology,1990,183:63-98.
[3] Dai Q,Li L H,Liu X Q,et al.Integrating overlapping structures and background information of words significantly improves biological sequence comparison[J].Plos One,2011,6(11):e26779.
[4] Bonham-Carter O,Steele J,Bastola D.Alignment-free genetic sequence comparisons:a review of recent approaches by word analysis[J/OL].Bioinformatics,2014,15(6):890-905.http://bib.oxfordjournals.org/content/15/6/890.
[5] Randic M,Zupan J,Balaban A T,et al.Graphical representation of proteins[J].Chemical Review,2011,111(2):790-862.
[6] 余宏杰.生物序列特征信息提取方法及其应[D].合肥:中国科学技术大学,2013.
[7] 邓伟.生物序列的相似性分析及K词模型研究[D].济南:山东大学,2015.
[8] Leimeister C A,Boden M,Horwege S,et al.Fast alignment-free sequence comparison using spaced-word frequencies[J].Bioinformatics,2014,30(14):1991-1999.
[9] Bao J,Yuan R,Bao Z.An improved alignment-free model for DNA sequence similarity metric[J].BMC Bioinformatics,2014,15(1):1-15.
[10] Li C,Wang J.Relative entropy of DNA and its application[J].Physica A Statistical Mechanics & Its Applications,2005,347(C):465-471.
[11] 许景皓.基于序列顺序信息的DNA结合蛋白质识别与远程同源性检测[D].哈尔滨:哈尔滨工业大学,2014.
[12] 杨希武.DNA序列比较的K-词非频率模型研究及应用[D].大连:大连理工大学,2013.
[13] 吴迪.基于加权相似度的序列聚类算法研究[D].燕山:燕山大学,2014.
[14] Shi L,Huang H.DNA sequences analysis based on classifications of nucleotide bases[C]//International Colloquium on Computing,communication,Control,and Management,2010:379-384.
[15] 黄玉娟.基于K词的DNA序列分析的模型研究及应用[D].大连:大连理工大学,2012.
[16] 胡清铭.蛋白质序列特征提取及其在亚细胞定位中的应用[D].长沙:湖南大学,2013.
[17] Kurgan L,Homaeian L.Prediction of Structural classes for protein sequences and domains-impact of prediction algorithms,sequence representation and homology and test procedures on accuracy[J].Pattern recognization,2006,39(12):2323-2343.
[18] 唐杰.基于K-字位置的新序列比较方法[D].杨凌:西北农林科技大学,2015.
[19] Wei D,Jiang Q,Wei Y,et al.A novel hierarchical clustering algorithm for gene sequences[J].Bmc Bioinformatics,2012,13(1):1-15.
[20] Wei D,Jiang Q.A DNA sequence distance measure approach for phylogenetic tree construction[C]//IEEE Fifth International Conference on Bio-Inspired Computing:Theories and Applications.IEEE,2010:204-212.
[21] HOGENOM:Database of complete genome homologous genes families[DB/OL].http://pbil.univ-lyon1.fr/databases/hogenom/home.Php.
ALIGNMENT-FREEMODELFORSEQUENCECLUSTERINGMETHODBASEDONLOCATIONINFORMATION
Wei Jing Xu Pengna Jiang Yu’e Lin Jie*
(FacultyofSoftware,FujianNormalUniversity,Fuzhou350108,Fujian,China)
Alignment-free similarity model for sequence calculates the similarity between the sequences by using the statistical information of the sequences, which has the advantage of fast calculation and high precision. Alignment-free model for sequence clustering method based on position information was proposed. The features of sequences can be obtained by combining the LF entropy of the corresponding word which was calculated from the Local Frequency of every word with the K-mers model, and the frequency of every word. This new method can be applied to protein clustering. The experimental results showed this new method improved the accuracy of clustering effectively.
K-mers Local frequency entropy Sequence clustering Position information
2017-01-23。国家自然科学基金项目(61472082);福建省自然科学基金项目(2014J01220)。魏静,硕士生,主研领域:数据挖掘。徐彭娜,硕士生。江育娥,教授。林劼,副教授。
TP3
A
10.3969/j.issn.1000-386x.2017.12.008
猜你喜欢
数学物理学报(2022年5期)2022-10-09 08:56:44
园林科技(2021年3期)2022-01-19 03:17:48
河北画报(2020年8期)2020-10-27 02:54:20
电子测试(2017年15期)2017-12-18 07:19:27
浙江大学学报(工学版)(2016年2期)2016-06-05 09:20:51
智能系统学报(2015年4期)2015-12-27 09:38:39
读者·校园版(2015年7期)2015-05-14 13:11:40
深圳大学学报(理工版)(2015年5期)2015-02-28 16:22:05
电子设计工程(2015年6期)2015-02-27 12:04:53
图书馆论坛(2014年8期)2014-03-11 18:47:59
