
HPLC法同时测定青娥丸中7种成分的含量
2017-12-13 09:49:18王向锋黄小为刘明霞朱玉青郯城县第一人民医院山东临沂7600山东罗欣药业集团股份有限公司山东临沂7607
中国药房 2017年33期
王向锋,黄小为,刘明霞,朱玉青(.郯城县第一人民医院,山东临沂7600;.山东罗欣药业集团股份有限公司,山东临沂 7607)
HPLC法同时测定青娥丸中7种成分的含量
王向锋1*,黄小为1,刘明霞2,朱玉青2(1.郯城县第一人民医院,山东临沂276100;2.山东罗欣药业集团股份有限公司,山东临沂 276017)
目的:建立同时测定青娥丸中补骨脂素、异补骨脂素、佛手柑内酯、欧前胡素、三甲基补骨脂素、新补骨脂异黄酮、补骨脂二氢黄酮含量的方法。方法:采用高效液相色谱法。色谱柱为Kinetex-C18,流动相为甲醇-0.1%乙酸溶液(梯度洗脱),流速为1.0 mL/min,检测波长为248 nm,柱温为30℃ ,进样量为20 μL。结果:补骨脂素、异补骨脂素、佛手柑内酯、欧前胡素、三甲基补骨脂素、新补骨脂异黄酮、补骨脂二氢黄酮检测质量浓度线性范围分别为1.012~101.2 μg/mL(r=0.999 9)、1.007~100.7 μg/mL(r=0.999 7)、1.010~101.0 μg/mL(r=0.999 9)、1.021~102.1 μg/mL(r=0.999 9)、1.002~100.2 μg/mL(r=0.999 6)、1.008~100.8 μg/mL(r=0.999 9)、1.025~102.5 μg/mL(r=0.999 8);定量限分别为0.15、0.15、0.30、0.30、0.15、0.30、0.30 μg/mL,检测限分别为0.05、0.05、0.10、0.10、0.05、0.10、0.10 μg/mL;精密度、稳定性、重复性试验的RSD<2.0%;加样回收率分别为95.3%~99.6%(RSD=1.9%,n=6)、96.1%~99.3%(RSD=1.2%,n=6)、95.2%~98.4%(RSD=1.4%,n=6)、95.4%~99.2%(RSD=1.5%,n=6)、96.1%~99.3%(RSD=1.5%,n=6)、95.6%~98.9%(RSD=1.4%,n=6)、95.2%~99.6%(RSD=1.6%,n=6)。结论:该方法简便、结果准确,适用于青娥丸中7种成分含量的同时测定。
高效液相色谱法;青娥丸;含量;补骨脂素;异补骨脂素;佛手柑内酯;欧前胡素;三甲基补骨脂素;新补骨脂异黄酮;补骨脂二氢黄酮
青娥丸具有补肾强腰的作用,主要用于肾虚腰痛、起坐不利、膝软乏力等症的治疗[1];由盐杜仲、盐补骨脂、大蒜(蒸熟粉碎成细粉)、核桃仁(炒后捣烂)4味中药组成[2-3]。盐补骨脂作为青娥丸的主要配方之一,具有温肾助阳、纳气平喘、温脾止泻的功效[4-7]。补骨脂中主要含有呋喃香豆素类、黄酮类、萜酚类成分[8-9]。青娥丸收载于2015年版《中国药典》(一部),其含量测定项中仅对补骨脂中补骨脂素和异补骨脂素两种呋喃香豆素类成分进行测定[1],并没有对其他功效成分进行控制;且现有关于测定青娥丸的文献多为单一成分报道[10-12]。鉴于此,本研究采用高效液相色谱法(HPLC)建立了同时测定青娥丸呋喃香豆素类成分(补骨脂素、异补骨脂素、佛手柑内酯、欧前胡素、三甲基补骨脂素)和黄酮类成分(新补骨脂异黄酮、补骨脂二氢黄酮)的含量,以期为完善该制剂的质量控制提供参考。
1 材料
1.1 仪器
1260型HPLC仪,包括G1329B自动进样器、G1315D二极管阵列检测器、G1311C四元梯度洗脱泵、G1316A柱温箱、1260色谱工作站(美国Agilent公司);XP205型十万分之一电子分析天平(瑞士Mettler-Toledo公司);Milli-Q型纯化水仪(美国Millipore公司);D101大孔吸附树脂(上海摩速科学器材有限公司)。
1.2 药品与试剂
青娥丸(南京同仁堂药业有限责任公司,批号:160301、160302、160303,规格:9 g/丸);补骨脂素对照品(批号:110739-201617,纯度:99.7%)、异补骨脂素对照品(批号:110738-201614,纯度:99.9%)、佛手柑内酯对照品(批号:520036-201401,纯度:99.2%)、欧前胡素对照品(批号:110826-201616,纯度:99.6%)均购自中国食品药品检定研究院;三甲基补骨脂素对照品(东京化成工业株式会社,批号:TYTUI-JF,纯度:98.0%);新补骨脂异黄酮对照品(批号:150528,纯度:98.0%)、补骨脂二氢黄酮对照品(批号:150415,纯度:98.0%)均购自北京北纳创联生物技术研究院;甲醇为色谱纯,乙酸为优级纯,其余试剂均为分析纯,水为纯化水。
2 方法与结果
2.1 色谱条件
色谱柱:Kinetex-C18(250 mm×4.6 mm,5 μm);流动相:甲醇(A)-0.1%乙酸溶液(B),梯度洗脱(洗脱程序见表1);流速:1.0 mL/min;检测波长:248 nm;柱温:30 ℃;进样量:20 μL。
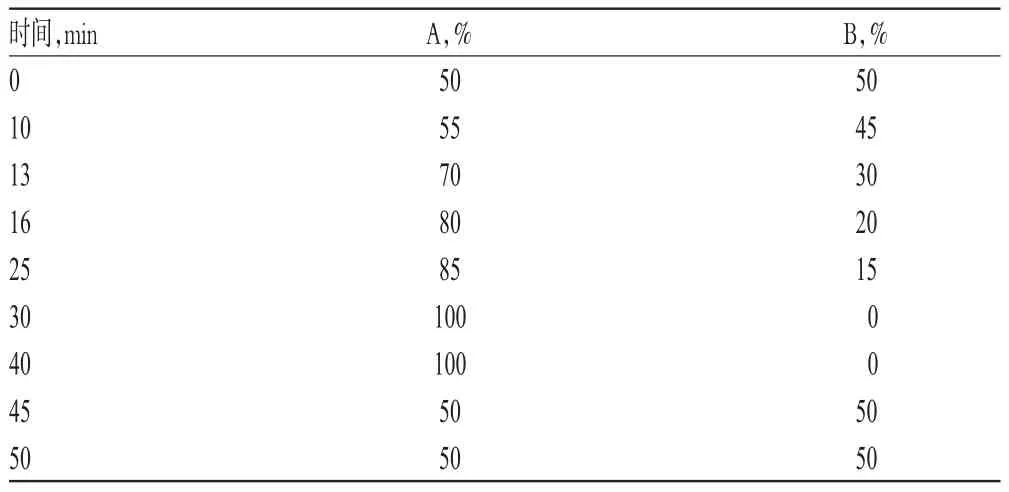
表1 梯度洗脱程序Tab 1 Gradient elution procedure
2.2 溶液的制备
2.2.1 混合对照品溶液 取待测成分对照品各适量,精密称定,置于同一50 mL量瓶中,加甲醇-0.1%乙酸溶液(1∶1,V/V)溶解并定容,摇匀,制成补骨脂素、异补骨脂素、佛手柑内酯、欧前胡素、三甲基补骨脂素、新补骨脂异黄酮、补骨脂二氢黄酮质量浓度分别为1.012、1.007、1.010、1.021、1.002、1.008、1.025 mg/mL的混合对照品贮备液。取上述混合对照品贮备液1 mL,置于10 mL量瓶中,加甲醇-0.1%乙酸溶液(1∶1,V/V)定容,摇匀,即得。
2.2.2 供试品溶液 取样品适量,研碎,精密称取约4.0 g,置于250 mL圆底烧瓶中,加甲醇50 mL,加热回流1 h,滤过,滤液蒸干,残渣加水10 mL使溶解,通过D101型大孔吸附树脂(内径为1.7 cm,柱高为15 cm),用30%乙醇溶液70 mL洗脱,弃去洗脱液,再用乙醇洗脱,收集乙醇洗脱液,蒸干,残渣加甲醇-0.1%乙酸溶液(1∶1,V/V)50 mL使溶解,滤过,取续滤液,即得。
2.2.3 空白对照溶液 取甲醇-0.1%乙酸溶液(1∶1,V/V)适量为空白对照溶液。
2.3 系统适用性试验
精密量取“2.2”项下混合对照品溶液、供试品溶液和空白溶剂各适量,按“2.1”项下色谱条件进样测定,记录色谱,详见图1。由图1可知,在该色谱条件下,各成分均能达到基线分离,分离度>1.5;理论板数以补骨脂素峰计为7 000,保留时间为7.2 min。结果表明,其他成分对测定无干扰[13-14]。
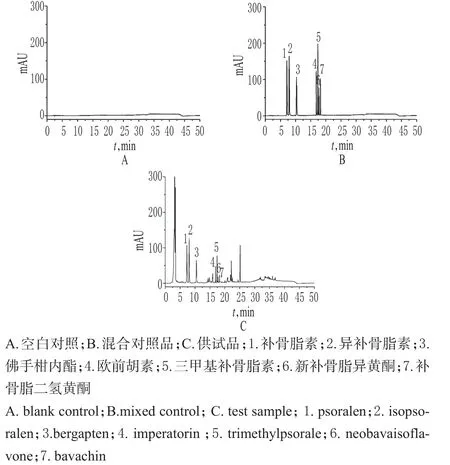
图1 高效液相色谱图Fig 1 HPLC chromatograms
2.4 线性关系考察
分别精密量取“2.2.1”项下混合对照品贮备液0.1、0.2、0.5、1、2、5、10 mL,分别置于100 mL量瓶中,加甲醇-0.1%乙酸溶液(1∶1,V/V)定容,摇匀,即得系列混合对照品溶液。取上述系列混合对照品溶液适量,按“2.1”项下色谱条件进样测定,记录峰面积。以待测成分质量浓度(x,μg/mL)为横坐标、峰面积(y)为纵坐标进行线性回归。回归方程与线性范围见表2。
2.5 定量限(LOQ)与检测限(LOD)考察
取“2.2.1”项下混合对照品溶液适量,倍比稀释,按“2.1”项下色谱条件连续进样测定6次,记录峰面积。当信噪比为10∶1时,得LOQ;当信噪比为3∶1时,得LOD,结果见表3。
2.6 精密度试验
精密量取“2.2.1”项下混合对照品溶液适量,按“2.1”项下色谱条件连续进样测定6次,记录峰面积。结果,补骨脂素、异补骨脂素、佛手柑内酯、欧前胡素、三甲基补骨脂素、新补骨脂异黄酮、补骨脂二氢黄酮峰面积的RSD分别为0.35%、0.38%、0.22%、0.56%、0.18%、0.47%、0.55%(n=6),表明仪器精密度良好。
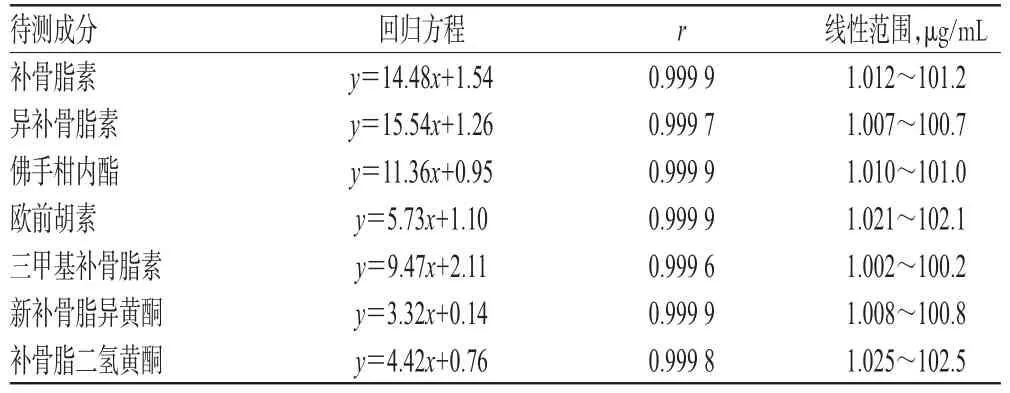
表2 回归方程与线性范围Tab 2 Regression equations and linear ranges
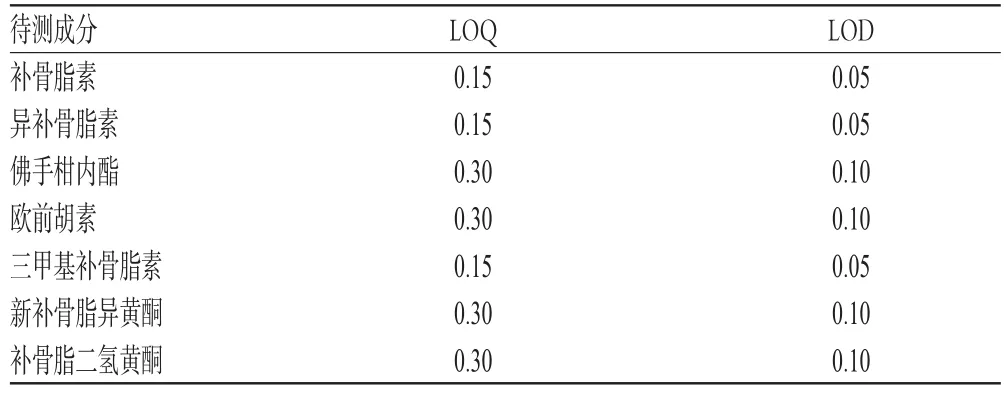
表3LOQ与LOD测定结果(μg/mL)Tab 3 Results of LOD and LOQ(μg/mL)
2.7 稳定性试验
取“2.2.2”项下供试品溶液(批号:160301)适量,分别于室温下放置0、2、4、8、12、24 h时按“2.1”项下色谱条件进样测定,记录峰面积。结果,补骨脂素、异补骨脂素、佛手柑内酯、欧前胡素、三甲基补骨脂素、新补骨脂异黄酮、补骨脂二氢黄酮峰面积的RSD分别为0.74%、0.82%、0.62%、0.75%、0.84%、0.62%、0.85%(n=6),表明供试品溶液在室温下放置24 h内稳定性良好。
2.8 重复性试验
取样品(批号:160301)适量,按“2.2.2”项下方法制备供试品溶液,共6份,按“2.1”项下色谱条件进样测定,记录峰面积并计算样品含量。结果,补骨脂素、异补骨脂素、佛手柑内酯、欧前胡素、三甲基补骨脂素、新补骨脂异黄酮、补骨脂二氢黄酮的含量平均值分别为0.811、0.746、0.647、0.650、0.774 、0.226 、0.368 mg/g;RSD分别为1.55%、1.21%、1.76%、1.65%、1.38%、1.52%、1.05%(n=6),表明本方法重复性良好。
2.9 加样回收率试验
精密称取同一批己知含量的样品(批号:160301)约2.0 g,共6份,各置于250 mL圆底烧瓶中,分别精密加入一定质量的待测成分对照品,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算加样回收率,结果见表4。
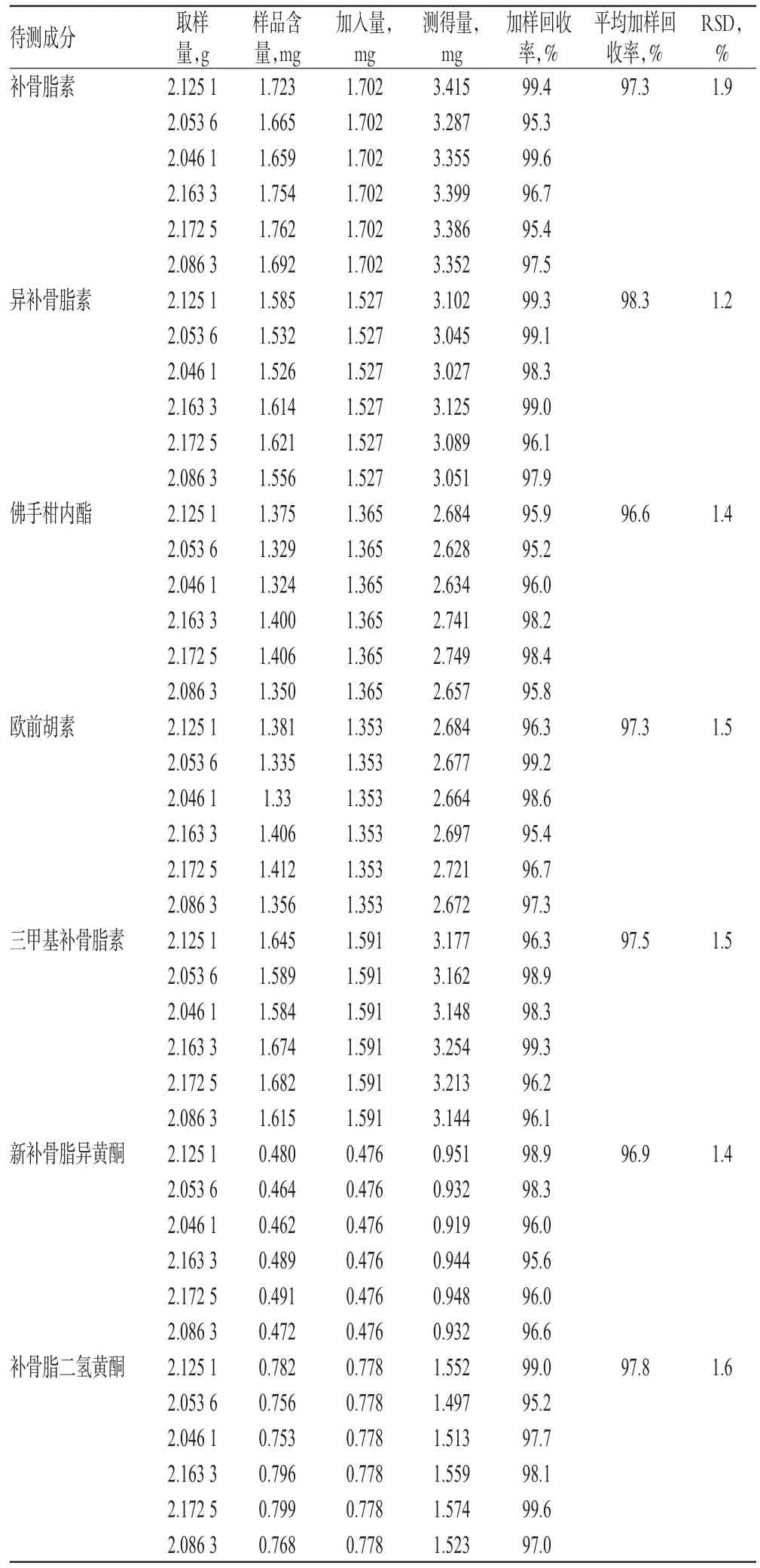
表4 加样回收率试验结果(n=6)Tab 4 Results of recovery tests(n=6)
2.10 样品含量测定
取3批样品各适量,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算样品含量,结果见表5。
3 讨论
3.1 样品提取方法的优点
本试验采用D101型大孔吸附树脂对样品进行富集处理,D101型大孔吸附树脂可对补骨脂中的呋喃香豆素类和黄酮类成分具有较强的吸附能力,采用30%乙醇溶液进行洗脱,可以将样品中极性较强的成分去除;然后采用乙醇进行洗脱,收集乙醇洗脱液,进行浓缩富集,有助于保护色谱柱和排除其他成分的干扰。此外,对样品进行富集,还有助于提高样品中微量成分的检出率。
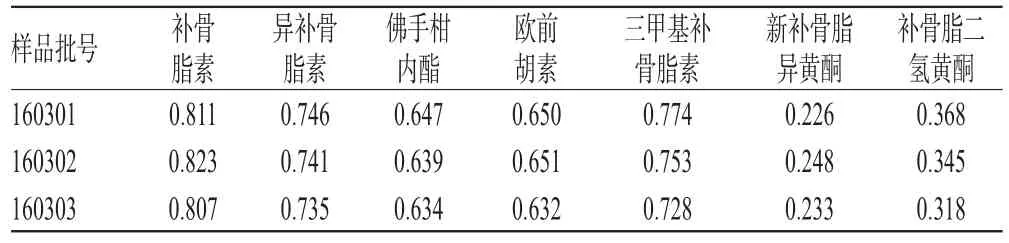
表5 样品含量测定结果(n=3,mg/g)Tab 5 Results of contents determination of samples(n=3,mg/g)
3.2 流动相的考察
本试验考察了甲醇-水、甲醇-0.1%乙酸溶液、甲醇-0.1%磷酸溶液作为流动相时进行梯度洗脱的色谱情况。结果,采用甲醇-水作为流动相进行梯度洗脱,色谱峰拖尾较为严重,且分离度不好;以甲醇-0.1%磷酸溶液作为流动相进行梯度洗脱,欧前胡素、三甲基补骨脂素、新补骨脂异黄酮与补骨脂二氢黄酮之间的分离度较差;以甲醇-0.1%乙酸溶液作为流动相进行梯度洗脱,待测成分均能较好分离。因此,选择甲醇-0.1%乙酸溶液为本试验的流动相。
3.3 检测波长的考察
笔者采用二极管阵列检测器在190~400 nm波长范围内进行扫描,发现待测成分在248 nm波长处均具有较强的紫外吸收,因此选择248 nm作为检测波长。
3.4 结果分析
2015年版《中国药典》(一部)规定,补骨脂素与异补骨脂素两种成分的总量应不得少于1.2 mg/g,但没有规定其他成分的含量。本试验测定结果中,补骨脂素与异补骨脂素两种成分的总量均能符合《中国药典》的规定。通过对3批样品进行含量测定,发现批次之间各成分的含量基本一致,说明企业的生产工艺稳定,批次之间样品的差异较小。
综上所述,本方法简便、结果准确,适用于青娥丸中7种成分含量的同时测定。
[1]国家药典委员会.中华人民共和国药典:一部[S].2015年版.北京:中国医药科技出版社:1024-1025.
[2]刘玲,翁泽斌,王恒,等.青娥丸方有效成分药动学-药效学相关性研究[J].中国中药杂志,2016,41(23):4436-4441.
[3]翁泽斌,颜翠萍,张志杰,等.不同炮制品入药的青娥丸含药血清对人成骨细胞增殖、分化及矿化的影响[J].中国实验方剂学杂志,2015,21(6):165-168.
[4]梁建军,徐亚莉,田树喜,等.补骨脂研究现状及前景[J].河北中医,2013,35(12):1904-1906.
[5]夏亚楠,余凌英,王德键,等.补骨脂不同炮制品对肾阳虚脾虚大鼠的影响研究[J].亚太传统医药,2016,12(9):5-7.
[6]辛丹,颜冬梅,王跃飞,等.补骨脂及其相关化学成分的药理与毒理研究进展[J].辽宁中医药大学学报,2009,11(7):70-72.
[7]邱蓉丽,李璘,乐巍.补骨脂的化学成分与药理作用研究进展[J].中药材,2010,33(10):1656-1659.
[8]颜冬梅,高秀梅.补骨脂化学成分研究进展[J].辽宁中医药大学学报,2012,14(9):96-99.
[9]吴疆,魏巍,袁永兵.补骨脂的化学成分和药理作用研究进展[J].药物评价研究,2011,34(3):217-219.
[10]张紫佳,陈洁,赵俊铭,等.超高效液相色谱法测定青娥丸中松脂醇二葡萄糖苷的含量[J].色谱,2010,28(8):805-808.
[11]陈洁,徐颖,张紫佳,等.青娥丸质量标准研究:君药杜仲的含量测定[J].中国实验方剂学杂志,2012,18(20):112-114.
[12]艾路,马群,邱落,等.高效液相色谱法测定加味青娥丸中补骨脂素、异补骨脂素的含量[J].世界科学技术·中药现代化,2010,12(5):808-810.
[13]王恒,李伟东,高倩倩,等.不同炮制品配伍的青娥丸中12种指标成分含量变化研究[J].中药新药与临床药理,2016,27(5):684-688.
[14]宋潇,戚爱棣,王跃飞,等.不同炮制方法对补骨脂中4类化学成分的影响[J].中国中药杂志,2011,36(15):2071-2075.
Simultaneous Determination of 7 Kinds of Components in Qing’e Pills by HPLC
WANG Xiangfeng1,HUANG Xiaowei1,LIU Mingxia2,ZHU Yuqing2(1.Tancheng County First People’s Hospital,Shandong Linyi 276100,China;2.Shandong Luoxin Pharmaceutical Group Co.,Ltd.,Shandong Linyi 276017,China)
OBJECTIVE:To develop a method for simultaneous determination of psoralen,isopsoralen,bergapten,imperatorin,trimethylpsorale,neobavaisoflavone and bavachin in Qing’e pills.METHODS:HPLC method was adopted.The separation was performed on Kinetex-C18column with mobile phase consisted of methanol-0.1%glacial acetic(gradient elution)at the flow rate of 1.0 mL/min.The detection wavelength was set at 248 nm.The column temperature was 30℃ .The sample size was 20 μL.RESULTS:The linear ranges were 1.012-101.2 μg/mL for psoralen(r=0.999 9),1.007-100.7 μg/mL for isopsoralen(r=0.999 7),1.010-101.0 μg/mL for bergapten(r=0.999 9),1.021-102.1 μg/mL for imperatorin(r=0.999 9),1.002-100.2 μg/mL for trimethylpsorale(r=0.999 6),1.008-100.8 μg/mL for neobavaisoflavone(r=0.999 9),1.025-102.5 μg/mL for bavachin(r=0.999 8),respectively.The limits of quantitation were 0.15,0.15,0.30,0.30,0.15,0.30,0.30 μg/mL,and the limits of detection were 0.05,0.05,0.10,0.10,0.05,0.10,0.10 μg/mL,respectively.RSDs of precision,stability and reproducibility tests were lower than 2.0%.The recoveries were 95.3%-99.6%(RSD=1.9%,n=6),96.1%-99.3%(RSD=1.2%,n=6),95.2%-98.4%(RSD=1.4%,n=6),95.4%-99.2%(RSD=1.5%,n=6),96.1%-99.3%(RSD=1.5%,n=6),95.6%-98.9%(RSD=1.4%,n=6),95.2%-99.6%(RSD=1.6%,n=6),respectively.CONCLUSIONS:The method is simple and accurate,can be used for simultaneous determination of 7 kinds of components in Qing’e pills.
HPLC;Qing’e pills;Content;Psoralen;Isopsoralen;Bergapten;Imperatorin;Trimethylpsorale;Neobavaisoflavone;Bavachin
R927.2
A
1001-0408(2017)33-4728-04
DOI 10.6039/j.issn.1001-0408.2017.33.33
*主管药师。研究方向:医药研发及管理。E-mail:wxf279@163.com
(编辑:刘 柳)
2017-04-18
2017-06-28)
猜你喜欢
中国农业大学学报(2023年5期)2023-05-11 13:26:58
天津中医药(2018年9期)2018-09-14 08:16:44
天然产物研究与开发(2018年5期)2018-06-13 03:23:42
中成药(2017年9期)2017-12-19 13:34:41
中成药(2017年6期)2017-06-13 07:30:35
中国民族民间医药(2014年17期)2014-09-11 09:38:42
中医研究(2014年4期)2014-03-11 20:28:42
中国中医药现代远程教育(2014年21期)2014-03-01 04:32:37
中成药(2014年10期)2014-02-28 22:29:30
中成药(2012年8期)2012-09-06 14:28:58
