
先天型白质消融性白质脑病1例及其基因突变分析
2017-11-29 03:06:51张晓莉王丽君贾天明
临床儿科杂志 2017年11期
张晓莉 王丽君 贾天明 韩 瑞 赵 鑫
郑州大学第三附属医院(河南郑州 450052)
先天型白质消融性白质脑病1例及其基因突变分析
张晓莉 王丽君 贾天明 韩 瑞 赵 鑫
郑州大学第三附属医院(河南郑州 450052)
目的探讨先天型白质消融性白质脑病(VWM)的临床及基因特点。方法回顾性分析1例先天型VWM患儿的临床资料及基因检测结果。结果患儿,男,3月龄,因间断抽搐2个月就诊,出生体质量1 900 g,生后有窒息史;发育落后,不能注视、追视,不能逗笑、抬头,下肢肌张力升高,双眼白内障。头颅CT及MRI可见大脑白质弥漫性异常,与脑脊液信号相同。基因检测结果显示,EIF2B5基因存在错义突变(c.1016G>A)和移码突变(c.1809delC),为复合杂合突变,其父母均为杂合子。结论VWM是遗传性白质脑病之一,先天型极为罕见,诊断需根据临床表现及EIF2B基因分析。移码突变位点c.1809delC国际上尚未见报道。
白质消融性白质脑病;EIF2B基因; 先天型
白质消融性白质脑病(leukoencepha1opathy with vanishing white matter diseas,VWM)是一种常染色体隐性遗传性白质脑病,1993年被首次报道,由编码真核细胞翻译启动因子2B(eukaryotic translation initiation factor 2B,ElF2B)的五个亚单位(α、β、δ、γ、ε)的基因EIF2B1-5任一突变所致[1,2]。白质消融性白质脑病主要表现为进行性的运动智力倒退,头颅MRI可见弥漫对称的白质异常,并随病情进展逐渐与脑脊液相同的信号。根据起病年龄及病程特点可分为先天型、婴儿型、早期儿童型、青少年型及成年型[3]。早期儿童型最常见[4],先天型国内尚未见报道。本研究回顾分析1例先天型VWM患儿的临床资料,及相关基因测序结果。
1 临床资料
患儿,男,3月龄,因“间断抽搐2个月,加重1周”入院。患儿系G1P1,胎龄37周经产道分娩,羊水Ⅲ°污染,出生体质量1 900 g,生后反应差,哭声弱。在当地医院诊断为“足月小样儿、新生儿窒息、新生儿缺氧缺血性脑病、肺炎”,住院治疗10天好转出院。出院后吃奶差、哭声弱。生后1个月在当地医院行眼底检查后诊断为“先天性白内障”,未治疗。生后1个月出现抽搐,表现为头部及四肢抖动,每日发作1~2次,每次持续数秒。入院前1周出现抽搐频繁,表现为眨眼,有时单侧,有时双侧,每次持续3~5分钟,间隔数分钟再次发作。当地医院给予补钙等治疗无效,遂来郑州大学第三附属医院。患儿父母体健,否认遗传性疾病史。入院体格检查:体温36.9℃,心率134次/min,呼吸34次/min,体质量3 000 g,头围33 cm;神志清,反应差,营养不良、贫血貌,哭声弱,前囟1.0 cm×1.0 cm,平软,双侧晶状体浑浊,口唇苍白,颈无抵 抗;心肺听诊无明显异常;腹软,肝右肋缘下1.5 cm,质软边锐,脾肋下未触及;无注视、追视,不能逗笑,俯卧位不能抬头,下肢肌张力升高。实验室检查:血常规血红蛋白73 g/L,余正常;血乳酸升高3.1 mmol/L;血气分析、血糖、血氨未见异常;甲状腺功能、电解质、脑脊液常规生化无异常。头颅CT示小脑齿状核对称性密度减低,双侧大脑白质弥漫性对称性低密度影,侧脑室对称性扩张(图1)。眼科会诊确诊双眼先天性白内障。动态脑电图,醒睡各期各导可见中高幅尖波、尖慢波发放。心脏彩超未见异常。考虑患儿为白质脑病可能性大,建议头颅MRI检查,家属未同意。住院期间予维生素B6,抽搐无减轻,先后加用丙戊酸钠口服液最大量30 mg/(kg·d);苯巴比妥针5 mg/(kg·d);托吡酯最大量5 mg/(kg·d);治疗效果差,仍有频繁抽搐。家属放弃治疗,住院16天自动出院。出院后3个月(6月龄)查头颅MRI示广泛大脑白质异常,T1Flair、T2及T2Flair像双侧大脑白质弥漫性异常,与脑脊液信号接近(图2)。患儿于7月龄夭折。

图1 患儿头颅CT
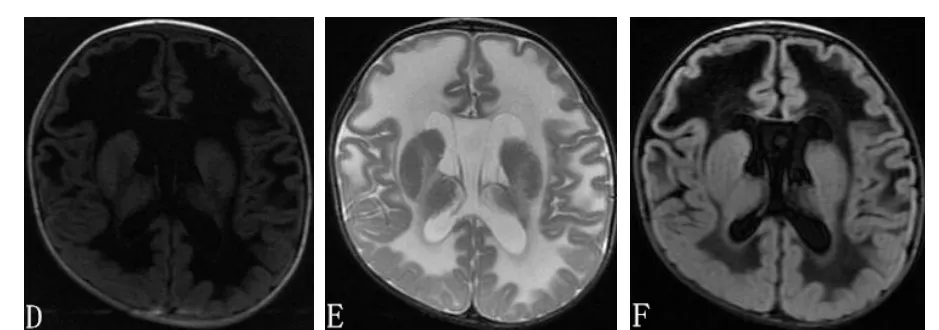
图2 患儿头颅MRI
结合患儿临床表现及头颅CT高度怀疑白质脑病,经医院伦理委员会审核,征得患儿父母的同意后,出院前抽取患儿及其父母的静脉血各2 mL,置于含乙二胺四乙酸抗凝试管中,送康旭医学检验基因公司进行二代测序(NEXTSEQ 500测序仪),原始数据经RTA software (real-timeanalysis,Illumina)、CASAVA software v1.8.2(Illumina)、BWA、Genome Analysis Toolkit(GATK) 生物信息学软件分析后,查找致病突变,并采用Sanger测序法对候选变异进行验证。基因检测结果示患儿EIF2B5基因存在以下突变:c.1016G>A,为错义突变,导致第339号氨基酸由Arg变为Gln(p.Arg339Gln);c.1809delC,为移码突变,导致从603号氨基酸Phe开始的氨基酸合成发生改变(p.Phe603fs),均为杂合子(图3)。c.1016G>A已经被证实与VWM有关[5],而c.1809delC致病性国内外尚未见报道。其父在位点 c.1016 发现 G>A 杂合变异,其母该位点未见异常;其母在位点 c.1809 发现 delC杂合性缺失,其父该位点未见异常,符合常染色体隐性遗传病突变位点家系分离规律。结合患儿发病年龄及临床表现,确诊为先天型VWM。

图3 患儿及其父母EIF2B5基因测序图
2 讨论
VWM又称儿童共济失调伴中枢神经系统髓鞘化不良,1998年由荷兰Vander Knaap正式命名此病,起病可从胎儿期至成人的任何年龄。Klingelhoefer等[4]总结了42篇文献中共283例患儿,其中有基因确诊的259例患儿中,先天型占2.7%,婴儿型3.1%,早期儿童型49%,青少年型占18.9%,成人型26.3%。国内报道29例基因阳性患儿,婴儿型10例(34%),早期儿童型16例(55%),青少年型3例(10%),无先天型病例[6]。
先天型VWM胎儿期即出现症状,如胎动减少、宫内发育迟缓、羊水减少,生后表现为喂养困难、呕吐、难治性癫痫、肌张力异常、低血糖、低体温等,脑白质异常同时伴有小头畸形、白内障、肾脏发育不良、卵巢发育不良等多器官异常,病情进展迅速,常在1岁内死亡[7]。婴儿型则为l岁内发病,常表现为肌张力低,随之出现惊厥、肢体痉挛、呼吸困难、呕吐、视力丧失、嗜唾、头围不增,多于2岁前死亡[3]。经典型即早期儿童型于l~5岁发病,多以运动功能倒退为主要表现,可逐渐出现共济失调、肢体痉挛、构音障碍、惊厥等,智力受累相对较轻;多于起病后l~5年死亡[3,4]。晚期儿童型及成人型起病相对隐匿,进展慢。该患儿为小于胎龄儿,宫内发育迟缓,出生时羊水污染,存在先天性白内障,生后发育迟缓、抽搐难以控制,7月龄即夭折,发病年龄及临床表现符合先天型VWM。VWM患儿病程中遇发热、轻微头部外伤等应激事件后发作性加重,疫苗接种可能导致病情进展[8],这点在儿童型患儿中更典型[4]。
VWM头颅MRI具有特征性改变,也是临床诊断的重要依据,可见弥漫性大脑白质广泛受累,累及中央区及皮层下白质,异常白质在Tl、T2及Flair像上逐渐演变为与脑脊液相同的信号[2]。小脑也可受累,表现为白质异常信号或萎缩,但通常不发生液化[7]。本例患儿生后3月龄头颅影像学可见双侧大脑半球脑白质弥漫性异常,并小脑齿状核异常信号,6月龄时查头颅MRI可见白质异常范围明显扩大,与报道相符。
EIF2B有 5个亚单位,任一亚单位的基因突变均可导致发病[2]。EIF2B5突变最为常见,国外报道占65%,国内仅为38%;而国内EIF2B3突变比例高达31%,明显高于白种人的4%;EIF2B1、2、4比例相当[9,10]。本例患儿为EIF2B5基因一个错义突变,一个移码突变,结合临床表现支持先天型VWM诊断。
突变的ElF2B分子如何引起发病目前尚不清楚。大脑白质区主要组成是神经元的突起及各种胶质细胞,尤其是星形胶质细胞及负责髓鞘形成的少突胶质细胞。VWM的病理表现:髓鞘缺失伴不同程度的轴索缺失,囊变区及周边部位少突胶质细胞明显减少[11]。利用RNAi技术在正常人类胶质前体细胞中敲除EIF2B5基因,抑制其基因表达,星形胶质细胞的诱导分化也严重受限[12]。研究发现,ElF2B突变使少突胶质细胞在基础状态即存在UPR通路的自身激活,内质网应激(ERS)后过度激活,导致少突胶质细胞的大量凋亡,进而推断减轻突变细胞的内质网负荷或稳定UPR的过度激活有望对疾病进展起到干预作用[13]。
VWM疾病严重程度及预后与发病年龄有关,越早发病预后越差[4]。先天型和婴儿型常在2岁前死亡,而成年人表现较轻且无特异性,常被忽视,表现为伴随有行为问题、痴呆等的精神异常[14,15]。基因型和临床表型之间的关系尚不明确。有文献报道,非保守区域氨基酸基因突变的患者发病年龄较晚、临床表现较轻、进展慢、生存时间长[4]。如常见的EIF2B5基因c.338G>A 突变不管是杂合子还是纯合子临床症状都较轻,而c.584G>A与先天型VWM有关,临床进展迅速[16]。 EIF2B-ε单位的p.Arg113His突变多见于合并有卵巢功能障碍的成年女性VWM患者,但个体差异性仍然很大,有携带该突变者4岁即发病的报道[12]。本例患儿发病早,进展快,预后差,推测与c.1809delC移码变异有关。
VWM是儿科中常见白质脑病之一,临床工作中,需要与球形细胞脑白质营养不良、X连锁肾上腺脑白质营养不良、线粒体脑肌病等疾病鉴别。本研究通过对此例先天型VWM患儿的临床资料和其家系EIF2B5基因分析,明确了诊断,为该家庭进行准确的遗传咨询提供了可能;发现一个新移码突变位点c.1809delC,丰富了EIF2B5基因突变谱。
[1]Hanefeld F,Holzhach U,Kruse B,et al.Diffuse white matter disease in three children:an eneephalopathy with unique features on magnetic resonance imaging and proton magnetic resonance spectroscopy [J].Neuropediatrics,1993,24(5):244-248.
[2]van der Knaap MS,Pronk JC,Scheper GC.Vanishing matter disease [J].Lancel Neurol,2006,5(5): 413-423.
[3]Valálik I,van der Knaap MS,Scheper GC,et al.Long-term tremor control with bilateral Vim-DBS in vanishing white matter disease [J].Parkinsonism Relat Disord,2012,18(9):1048-1050.
[4]Klingelhoefer L,Misbahuddin A,Jawad T,et al.Vanishing white matter disease presenting as opsoclonus myoclonus syndrome in childhood--a case report and review of the literature [J].Pediatr Neurol,2014,51(1): 157-164.
[5]Leegwater PA,Vermeulen G,Könst AA,et al.Subunits of the translation initiation factoreIF2Bare mutant in leukoence phalopathy with vanishing white matter [J].Nat Genet,2001,29(4): 383-388.
[6]张海华,吴晔,陈娜,等.中国白质消融性白质脑病患儿EIF2B1-5基因型特点[J].中华实用儿科临床杂志,2014,29(1): 52-56.
[7]van der Knaap MS,van Berkel CG,Herms J,et al.eIF2B-related disorders: antenatal onset and involvement of multiple organs [J].Am J Hum Genet,2003,73(5): 1199-1207.
[8]Takano K,Tsuyusaki Y,Sato M,et al.A Japanese girl with an early-infantile onset vanishing white matter disease resembling Cree leukoencephalopathy [J].Brain Dev,2015,37(6):638-642.
[9]Pronk JC,van Kollenburg B,Scheper GC,et al.Vanishing white matter disease: a review with focus on its genetics [J].Ment Retard Dev Disabil Res Rev,2006,12(2): 123-128.
[10]Zhang H,Dai L,Chen N,et al.Fifteen novelEIF2B1-5 mutations identified in Chinese children with leukoencephalopathy with vanishing white matter and a long term follow-up [J].PLoS One,2015,10(3): eoll8001.
[11]Bugiani M,Boor I,Powers JM,et al.Leukoencephalopathy with vanishing white matter: a review [J].J Neuropathol Exp Neurol,2010,69(10): 987-996.
[12]Dietrich J,Lacagnina M,Gass D,et al.EIF2B5 mutations compromise GFAP+ astrocyte generation in vanishing white matterleukodystrophy [J].Nat Med,2005,11(3): 277-283.
[13]陈娜,代丽芳,张海华,等.未折叠蛋白反应通路持续过度激活在白质消融性白质脑病发病中的作用[J].中华实用儿科临床杂志,2015,30(12): 895-899.
[14]Woody AL,Hsieh DT,McIver HK,et al.Infantile onset vanishing white matter disease associated with a novelEIF2 B5 variant,remarkablylong life span,severe epilepsy,and hypopituitarism [J].Am J Med Genet A,2015,167(4):826-830.[15]Labauge P,Horzinski L,Ayrignac X,et al.Natural history of adult-onseteIF2B-related disorders: a multi-centric survey of 16 cases [J].Brain,2009; 132(Pt 8): 2161-2169.
[16]Turón-Viñas E,Pineda M,Cusí V,et al.Vanishing white matter disease in a spanish population [J].J Cent Nerv Syst Dis,2014,13(6): 59-68.
2017-05-05)
(本文编辑:梁 华)
The clinical and genetic features of a case with antenatal form of leukoencephalopathy with vanishing white matter disease
ZHANG Xiaoli,WANG Lijun,JIA Tianming,HAN Rui,ZHAO Xin(The Third Affiliated Hospital of Zhengzhou University,Zhengzhou 450052,Henan,China)
ObjectiveTo explore the clinical features and gene mutations of antenatal form leukoencepha1opathy with vanishing white matter disease (VWM).MethodsThe clinical data and genetic test results in a patient with antenatal form of VWM were retrospectively analyzed.ResultsA three months old patient was admitted to our hospital with intermittent convulsions commenced from the first month after birth.The baby had low birth weight (1900g) and asphyxia at birth.Developmental retardation and cataracts in both eyes were found on physical examination,and the patient couldn’t stare,gaze-following,be amused and raise his head.In addition,he showed hypermyotonia of both lower extremities.Diffused and symmetrical abnormal signals same as that of the cerebrospinal fl uid in the cerebral white matter were observed by brain CT and MRI scanning,and the lesions were gradually enlarged.Moreover,a missense mutation (c.1016G>A) and a frameshift mutation(c.1809delC) inEIF2B5gene inherited from his parent were detected by DNA sequencing.ConclusionsVWM is one of the most prevalently inherited childhood white matter disorders,but the case of antenatal form is very rare.The diagnosis should be based on clinical manifestations andEIF2Bgenetic analysis.To our knowledge,the frameshift mutation c.1809delC has never been reported to date.
leukoencepha1opathy with vanishing white matter disease;ElF2Bgene; antenatal form
10.3969/j.issn.1000-3606.2017.11.002
贾天明 电子信箱: jtm226@sina.com
猜你喜欢
湘潮(上半月)(2022年8期)2022-12-12 03:45:28
大观(2018年8期)2018-01-23 18:02:37
山西大同大学学报(自然科学版)(2016年2期)2016-12-12 03:19:27
河北医学(2016年5期)2016-12-01 03:58:56
实用临床医学(2016年8期)2016-06-07 01:28:23
扬子江(2016年1期)2016-05-19 23:29:21
中华老年多器官疾病杂志(2016年8期)2016-05-14 07:17:02
河北中医(2016年12期)2016-03-08 06:04:54
西南军医(2015年1期)2015-01-22 09:08:31
河北医科大学学报(2010年10期)2010-03-25 10:14:56
