
钙钛矿反铁电氧化物的研究进展
2017-11-02 03:19:13冯玉军庄永勇魏晓勇
物理学进展 2017年5期
田 野,靳 立,冯玉军,庄永勇,徐 卓,魏晓勇
电子陶瓷与器件教育部重点实验室,国际电介质研究中心,西安交通大学,710049,西安
钙钛矿反铁电氧化物的研究进展
田 野,靳 立,冯玉军,庄永勇,徐 卓,魏晓勇∗
电子陶瓷与器件教育部重点实验室,国际电介质研究中心,西安交通大学,710049,西安
反铁电材料,因其电场诱导的反铁电–铁电相变行为,而在当代电子技术中有着潜在的应用价值。然而,自然界中存在的反铁电材料是相当稀缺的,人们对它的认识与理解有限,其亚晶格极化、有序结构、局域结构及其对性能的影响依然是目前学术争辩的热点话题。为应对全球日益剧增的大气污染状况和人类健康所面临的潜在威胁,“环境友好”的无铅材料已然成为近些年来全球科学家与工程师们关注的焦点。本文介绍了“反铁电”的概念、定义、结构与电学性质特点以及反铁电体的应用。综述了自1951年“反铁电”概念被提出以来,人们在钙钛矿结构中陆续发现的反铁电氧化物及其晶体结构和电学性质的研究进展,从晶格动力学角度对钙钛矿氧化物反铁电性起源问题做了评论。期望通过微观结构设计的角度为无铅反铁电材料的开发提供新的思路。
无铅;反铁电;钙钛矿结构
目 录
I.引言 155
II.反铁电的概念、定义及其基本电学性质 156
A.反铁电的概念以及反铁电体的定义 156
B.反铁电体的基本电学性质 157
III.反铁电材料的主要应用 157
A.高功率储能电容器 157
B.大位移致动器和水声换能器 159
C.爆电换能器 159
D.红外热释电探测与电卡制冷 160
IV.钙钛矿金属氧化物 161
A.铁电扭曲 162
B.反铁扭曲 162
V.反铁电的钙钛矿氧化物及其研究进展 164
A.锆酸铅与铪酸铅 164
B.铌酸钠 165
C.铌酸银 167
VI.具有反铁电扭曲的钙钛矿固溶体氧化物及其研究进展 168
A.钛酸锶钙 168
B.稀土离子取代的铁酸铋 169
C.钛酸铋钠和钛酸铋钠-钛酸钡固溶体 171
VII.钙钛矿金属氧化物中的反铁电性起源 174
VIII.全文总结与展望 176
A.全文总结 176
B.展望 177
致 谢 177
177
I.引言
在功能电介质材料中,特别是铁电(Ferroelectrics,简称 FE)和反铁电(Antiferroelectrics,简称AFE)材料,因其在外部因素 (如温度、电、光、力等)的作用下,表现出独特的物理性质,而成为众多光电信息功能电子元器件中必不可少的关键材料[1]。然而,当前商业使用的FE和AFE材料多以含铅化合物(如:锆钛酸铅,简称PZT)为主,而这类材料中所使用的原料-氧化铅(或者四氧化三铅)约占原料总质量的70%,它们在高温生产过程中容易挥发,而导致大气中“铅”含量的升高[2]。众所周知,人体中的微量元素“铅”超标,轻者会引起神经衰弱,重者会出现贫血、腹痛,严重者则会导致中毒性脑病。早在上个世纪后期,“铅”已经被列为具有“毒”性的重金属元素之一。近些年来,随着世界各国对大气环境保护的重视以及各国人民对人类可持续发展的强烈渴望,欧盟公布了《关于报废电子电器设备指令》(WEEE)和《关于在电子电器设备中禁止使用某些有害物质指令》(ROHS)。我国信息产业部也制定了《电子信息产品污染防治管理办法》。《办法》要求自 2006年 1月 1日起,列入电子信息产品污染重点防治目录的电子信息产品中不得含有铅、汞、镉、六价铬、聚合溴化联苯、聚合漠化联苯乙酸及其它有害物质[3,4]。研究和开发无铅电介质功能材料已然成为当下全球科学家、工程师们关注的焦点。近些年来,人们在无铅铁电领域,尤其是对无铅压电材料的探索,已取得了瞩目的成就[5]。这些成果不仅推动着材料科学的发展,也使无铅材料未来能够走进人们的生活迈进了一大步。然而,与热门的铁电材料相比较,人们对反铁电材料,特别是无铅反铁电材料的认识与理解却显得进展缓慢,这个特点充分反映在近十几年来检索的文献数量上(如图1所示)。

图1.Web of science数据库中检索的从2000年~2016年的文献数量(备注:主题检索“lead-free ferroelectric”、“leadfree piezoelectric”和“lead-free antiferroelectric”)
II.反铁电的概念、定义及其基本电学性质
A.反铁电的概念以及反铁电体的定义
非中心对称晶体中,具有自发极化,且自发极化方向因外电场方向反向而反向,称之为铁电性(Ferroelectricity),而具有铁电性的晶体则被称之为铁电体(Ferroelectrics,简FEs)。因此,铁电体同时是热释电体也是压电体。类比于铁电体,1951年,美国物理学家C.Kittle首次通过简单的“双子晶格模型”提出了反铁电(Antiferroelectric,AFE)的概念。基本思想是:晶格中,相邻子晶格中的阳离子彼此自发极化地沿着相反的方向,而整个晶格宏观上不显示净极化强度(如图2所示),并从宏观唯象理论的角度预言了反铁电晶体 (Antiferroelectrics)的存在[6]。

图2.反铁电体和铁电体的相邻晶格离子自发极化示意图[7]
然而,关于反铁电体的定义,随着更多化合物的发现以及对这些化合物的研究结果来看,目前确是比较微妙的,缺乏一个标准。从晶格动力学角度,M.E.Lines和A.M.Glass在1977年出版的标准参考书中这样写道:“反铁电体是一种由非极性软模冻结而形成的一种相,该相在转变温度附近具有大的介电响应。并且该相在电场作用下,可以转变为铁电相[8]。而在2005年出版的L.Bornstein编写的《铁电体与反铁电体》概述中更强调反铁电性[9],他提出:“反铁电晶体是这样一种晶体,它的结构是由自发极化方向完全相反的两种亚晶格所组成,并且该结构在电场下能转变成铁电相。自发极化是反铁电体所必要的,但对于反铁电性来说,它不是充分条件。反铁电性的概念,不仅体现在晶体结构上,而且体现在晶体的介电行为上。”早在锆酸铅的研究中,日本学者G.Shirane等人[10]根据“双子晶格模型”,从晶体结构与热力学自由能两方面考虑,提出了这样一个定义:“反铁电体是一种反极性晶体,它的自由能与具有自发极化的子晶格的极性晶体相当,并且在电场作用下,能够转变成铁电相。反铁电相与铁电相存在竞争,是反铁电体的内部特征。”最近,K.M.Rabe[11]这样描述到:“一个反铁电体就像铁电体,它的晶体结构是由一个具有高对称性的非极性相畸变(即,高对称性的非极性相转变为对称性较低的非极性相)后得到;对铁电体来说,这种畸变是极性的(即,高对称性的非极性相转变为对称性较低的极性相),而对于反铁电体来说,这种畸变是非极性的。但是,不是所有经过非极性畸变后得到的非极性相是反铁电体,必须有一个经过同样的高对称性的非极性相发生极性畸变后保持较低能态的铁电相与之对应,并在电场作用下,该铁电相能够被诱导通过一级相变。”该描述,从热力学、晶格动力学以及晶格对称性角度,给如何定义反铁电体提出了一个更加明晰的范畴。
B.反铁电体的基本电学性质
虽然阐述反铁电体的精确定义,目前学术界尚存争议。但是,反铁电体所具有的基本电学性质普遍一致,没有争议。一个区别线性介电体与反铁电体的重要依据就是双电滞回线(如图3(a)所示)。当施加电场低于反铁电转变为铁电相(简称AFE→FE)的相变场强(简称EF)时,晶体表现出线性介电体的性质。而当施加电场高于EF时,极化值突然跳跃到一个非常高的水平并接近饱和。此时,晶体表现出典型铁电体极化的性质。而当撤销电场或减小电场到场诱导的铁电相转变成反铁电相(简称FE→AFE)的关键电场(简称EA)时,场诱导的铁电相返回到反铁电相。而在零场时,晶体为反铁电相。当施加反向电场时,电极化响应出现相似的特征。而在整个正负交替的交流电场下,表现出双电滞回线的特征。当AFE↔FE相变发生时,除了极化值的突变外,晶胞体积也随之发生变化。一般,当 AFE→FE相变发生时,晶胞的体积膨胀。而逆相变发生时,晶胞体积收缩。该可逆过程,伴随着一个显著的应变突跃,从而表现出大的电致伸缩系数。因此,在正负交替的交流电场下,表现出与铁电体完全不同的电致应变滞后回线(如图3(b)所示)。

图3.反铁电体的双电滞回线和电致应变曲线
另一个区分线性介电体与反铁电体的性质就是介电行为。由于AFE概念的提出是基于人们对铁电晶体的认识。因而,反铁电体也应当具有与铁电体类似的介电行为。当反铁电体的晶格中程自发的反平行排列的离子位移极化,在接近相变温度时突然消失,同时伴随着大的介电异常峰(如图4所示)。此时,在高于相变温度的顺电相(简称PE)区域,介电常数服从如公式(1)所示,的居里–外斯(Curie-Weiss)定律。

式中:C为居里常数;T0是居里–外斯温度或特征温度;ε为介电常数

图4.反铁电体的相对介电常数εr与温度的关系
III.反铁电材料的主要应用
由于反铁电材料所具有的电场诱导的AFE-FE相变行为,表现出一些优越的电学性质,因而在某些应用领域具有潜在开发与研究的价值。当然,这些应用都是基于对钙钛矿铅基反铁电材料的应用探索。
A.高功率储能电容器
介电电容器,与化学电池,超级电容器相比,具有高的功率密度 (~GW/kg)、非常长的循环次数 (>106)、以及充电和放电速度快(~ns)的特点。特别适合作为需求高功率的脉冲电子器件,进而应用于军事装备如:电热激发炮、轨道炮、高能护盾、激光聚变系统等,医疗设备如:心脏除颤器、激光手术等。近年来,介电电容器在光缆损伤探测、电动汽车、石油天然气勘探等领域的应用也成为研究热点[12]。
一个典型的介电电容器是由两个导电的平板中间填充介电材料所组成。存储电能是电容器最基本的功能。其基本原理是当对电容器通电时,由于电容器两边的电势差,从而使得导电平板间填充的电介质中的正负电荷以相反的方向发生迁移而积累在导电的平行板上(如图5所示),这个过程被称之为充电过程,而存储的电能可以用下面的公式来定量:

式中:U是存储的电能;V为施加在电容器上的电压;Q为导电平行板上积累的电荷;C为电容器的电容。而对电介质来说,在电场的作用下,发生了电位移,其位移量D(D=ε0εrE)等于积累在导电平行板上的电荷密度(Q/A),A为导电的平行板的面积。因此电容器的电能存储密度W可以用下面的方程来表达:

公式中:E为电场,它的大小等于V/d。该公式只适用于一般线性介电材料。而对于高介电材料如铁电体来说,D=ε0εrE+P,其中P为铁电体的自发极化,D≈P。而对于反铁电体来说,如前所述,由于其低场下保持了线性介电体的性质。高场作用下,由于发生AFE↔FE相变时,D值突然猛增到一个非常高的水平并接近饱和,而该值等于电场诱导的铁电相的自发极化。因而,反铁电体表现出非常高的能量存储密度,与其他两种介电材料相比,特别适合作为需求高能量密度的介电电容器。如图6所示,比较了三种介电体的电极化测试曲线。从图中可以看出,与一般介电和铁电材料比较,反铁电材料显示了更高的W。

图5.平板电容器示意图[12]
通常情况下,AFE和FE材料由于在第一次施加电场之后,一部分能量存储在材料内部,而不能释放出来,如图7所示。因此,FE和AFE材料可释放能量存储密度Wrec的计算公式可表达为:

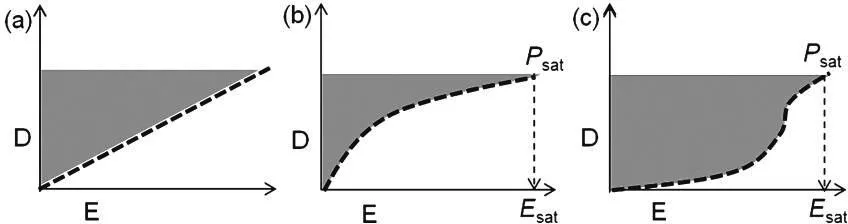
图6.介电材料电能存储密度[13]:(a)线性介电体,(b)铁电体,(c)反铁电体。图中,E: 电场。D:电位移;Psat:铁电饱和极化;Esat:饱和电场。灰色区域体现了储存的电能密度W。
式中:Pmax为最大极化强度;Pr为剩余极化强度。E为电场强度。
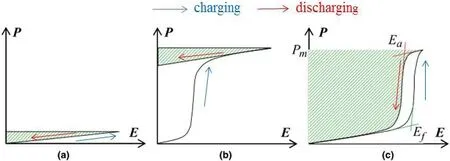
图7.三种介电材料的电滞回线和可持续使用的充放电示意图(a)线性介电材料(b)铁电材料(c)反铁电材料(图中的阴影面积体现了材料可释放的储能密度)
为了更加具体的表述反铁电体具有优越的能量存储特性,图8列出了笔者在发表的AgNbO3基固溶体的文章中[14],总结的目前发展的具有代表性的几种无铅陶瓷材料(包括反铁电、铁电以及顺电体)有效储能密度Wrec与应用电场E的关系,并划分出他们的性能区域。可以看出具有AFE特征的陶瓷材料,显示出较高的Wrec。

图8.当前发展的无铅陶瓷材料的储能特性与应用场强的关系(BT-based:钛酸钡基;BNT-based:钛酸铋钠基;AN:铌酸银;ABN:铋改性的铌酸银;KNN-based:铌酸钾钠基)[14]
B.大位移致动器和水声换能器
大位移致动器需求材料在较小的驱动电压下,就可以产生较大的位移量。当反铁电材料在发生电场诱导的AFE→FE相变时,除了极化值陡增到一个非常高的水平外,同时也伴随着材料的晶胞体积突然膨胀。在膨胀过程,材料内部会产生非常高的形变量,特别是在材料的纵向上。通过微观结构设计的手段,降低AFE→FE相变所需场强,使其在较低的电场驱动下,便可以诱导AFE相转变为FE相。在单晶样品中其形变量(S)高达1%。而在多晶陶瓷以及薄膜样品中,其形变量依旧维持在0.3~0.6%的水平(如图9所示)[15]。该性能远远高于于传统的压电材料和电致伸缩材料。而利用AFE材料制作的驱动器除过位移量大以外,并且响应灵敏、制作成本低。关于利用AFE-FE相变过程中产生大的应变的这一特性而制作水声换能器的报道并不多见。然而,早在1968年,Berlincourt就分析和探讨了使用AFE材料制作水声换能器的可行性并指出,利用AFE材料制作的换能器具有结构简单、尺寸小以及重量轻等特点[16]。中国声学所的科研人员采用AFE陶瓷首次开发了纵振式水声换能器。与传统的利用压电材料制作的换能器相比较,AFE水声换能器等效发射灵敏度提高了3~5 dB[17]。

图 9.锆钛锡酸铅镧 (PLZST)反铁电陶瓷电场强度 (E)与应变 (S)的关系 [15],Volume:体积;Longitudinal:纵向;Transverse:横向;
C.爆电换能器
铁电体器件在外加直流电场中进行极化时,其电畴取向趋向外电场方向。当外电场撤除后,电畴将保留一定的定向排列而形成剩余极化。同时,在电极上保留被剩余极化所束缚的电荷,这就意味着已有静电能贮存于铁电体内部。当爆炸形成的冲击波通过铁电体时,在冲击波压力的作用下,电畴被打乱、破坏或解体,剩余极化消失,电极上的束缚电荷变成自由电荷,这些电荷再通过负载向外输出电能从而实现能量的转换。然而,极化后的铁电体在冲击波或等静压力作用下实现能量转换所需的压力非常高约在GPa数量级[18]。与铁电体作为爆电换能的工作原理相似,通过微观结构设计的方法如:掺杂、改性等手段,从而使得AFE材料在施加较低的电场强度下,便能转变成FE态。然而,在电场撤销后,场诱导的FE态并不能返回到AFE状态,而以亚稳定的方式存在(如图 10(b)所示)。

图10.(a)反铁电的双电滞回线显示了可逆的AFE→FE相变(b)初始单圈电滞回线显示了不可逆的AFE→FE相变。Ef:AFE→FE 相变电场;Ea:FE→AFE 相变电场;Ec:铁电态的矫顽场。
通过适量的等静压或爆炸冲击波压力作用下(通常在 MPa数量级,显著低于铁电体),处于亚稳定的FE态能够转变到反铁电态。在此过程中,瞬间释放出所贮存的能量,产生具有非常高功率的电脉冲。图11阐述了一个理想化的等静压力驱动相变的能量收集模型。在开放电路中,当施加等静压时,电位移D将一直保持常数。因为FE体的D=ε0εrE+P当极化P降低到电位移不在改变时,电场必须增加而保D值不变(此时路径,如图11中的水平向右箭头所示)。该模型反映了一个非线性电介质在等静压力驱动的相变过程中的充电过程。接着对该介质连接负载时,电场降为0,存储的电荷释放完,该过程为放电过程。施加等静压程度不同,充电的路径与大小也有区别。
当施加等静压的程度只在FE-AFE转变所需的等静压力以上(如图 11所示),该路径只包括了 AFE相的撤除负载的部分,充电过程包括了FE-AFE转变和AFE相的线性电位移部分(如图11中箭头所示),存储的能量为阴影区域的面积。当施加更高的等静压时,电场驱动的AFE-FE和FE-AFE转变所需的电场强度转移到更高值。此时,最大能量密度为该电场下,AFE相的线性电位移曲线与FE体的剩余极化Pr所包围的三角区域的面积(如图9中虚线所包围的三角区域)。因此,要想获得大的能量密度,AFE材料的本征介电常数要足够小,而场诱导的FE相的剩余极化Pr要足够大。如此,三角形区域的面积才足够大。因此,理论上的能量密度W公式可以表达为[19]:


图11.电场诱导的FE态在压力与电场下的介电行为,图中虚线所包围的三角区域,体现了在等静压力作用下,可以释放的最大能量密度[19]
式中:Pr为诱导的FE相的剩余极化,εrAFE为AFE的相对介电常数。
D.红外热释电探测与电卡制冷
热释电效应主要涉及到晶体中极化随温度变化的特性。在加热和冷却过程中,材料的晶体结构中的原子位置发生改变,而影响到材料的极化,从而产生电压信号。热释电性能可以通过热释电系数p和和热释电材料综合性能的探测率优值FD来衡量[20]。它们的计算公式如下:

式中:D是电位移;T是温度;E是电场;PS是自发极化;ε0是真空介电常数;εr是相对介电常数;Cv是热容;tanδ是介电损耗。热释电红外探测器主要分为两种工作模式:一种是以锆钛酸铅(PZT)等铁电材料为主的本征热释电模式。该模式中由于热释电系数、介电性能受温度影响极大,使得探测器工作性能稳定性较差。另一种是以钙钛酸钡(BZT)和钛酸锶钡(BST)等弛豫铁电材料为代表的热释电–介电工作模式。该模式中,材料的Tm(最大介电常数所在温度)与工作温度相近,在加偏置电压的情况下,Tm附近材料的介电常数易受偏置电压的影响,对材料的热释电信号会有额外的贡献,而成为场致增强模式。同第一种模式相比较,由于工作在这种模式中的样品不需要极化,其的工作温度范围可以扩展到Tm温度附近且具有更高的灵敏度。同时,在这一模式中材料的介电常数和热释电系数的峰值及峰位会随着偏置电场的大小发生移动,使得热释电响应在一定温度范围内具有可调节的性能。场致热释电系数的计算公式可以表达为[21]:

式中:ε0和εr分别是真空和相对介电常数;E为外加直流电场;p0为零场下材料的本征热释电系数。BST和BZT这类弛豫铁电材料,由于PNRs的存在,电场作用下Tm附近介电常数和介电损耗随着电场强度增加而逐渐降低,而Tm介电峰出现展宽。这些特点导致介电常数随温度的变化率(dεr/dT)降低。当超过某个电场后,热释电系数p反而降低不如低电场的情况,对热释电性能的改善有限。反铁电体宏观上并不表现出极化特性,因而不具有热释电效应。然而,在电场作用下可以诱导晶体结构从AFE态向FE态转变,而表现出热释电性。在加热过程中,不同于先前的弛豫铁电材料,在场致模式下,AFE-PE转变处的介电峰得到增强,而介电损耗却降低。增强的介电非线性,意味着场致模式下介电常数随温度的变化率(dεr/dT)提高,从而显著提高材料的热释电系数p。而与弛豫铁电材料(BST和BZT)相比,AFE材料通常具有较小的相对介电常数εr,因而具有更高的探测优值 (FD)。人们对锆酸铅为基的AFE固溶体材料的热释电特性的研究中发现,通过调节偏置电场,还可以改变热释电流峰的温度以及热释电性能(如图12所示)。因而,AFE材料在场致热释电-介电工作模式下的红外探测有着显著的优势,为制作高灵敏度的红外探测敏感元件提供了可能。
电卡效应是热释电效应的逆效应。在绝热状态下,对材料施加电场或者撤销电场,从而使得温度发生改变。电卡效应的原理可以被解释为当对材料施加电场,材料微观组织结构变的更加有序,从而减小了极化熵。如果在绝热状态下,这个过程发生的话,总共的熵应该不变。因而,温度会增加从而补偿因电极化而减小的熵值。相反的,当撤销电场时,材料的微观组织结构回到混乱状态,材料的温度必须降低从而补偿增加的极化熵。利用这一原理,可以制作制冷器件。与蒸汽压缩制冷的原理相似,电卡制冷同样遵从卡诺循环的每个阶段。只不过,电卡制冷的媒介是固体并且不会产生温室效应。因而,成为新型的制冷技术[23,24]。根据 Maxwell关系:(∂D/∂T)E=(∂S/∂E)T,在施加电场的情况下,材料的温度改变(ΔT)和熵变(ΔS)可以表达为[25]:

表I.PLZST反铁电陶瓷的能量收集性能[19]

图12.Pb0.97La0.02(Zr0.75Sn0.09Ti0.16)O3AFE陶瓷在施加不同程度D.C偏置电场下的热释电电流和热释电系数随温度变化的曲线[22]

式中:ρ是质量密度;C是热容;T是温度;D是在最大电场下的电极化;E1和E2分别是起始电场和终止电场;其中就是热释电系数。根据公式可见,要想获得大的ΔT和ΔS,材料必须具有高的热释电系数。通常情况下,材料在发生相变的过程中,会产生大的热释电系数。对铁电体而言,热释电系数在FE-PE相变时达到峰值。而反铁电体,在偏置电场下成为铁电体,而场诱导的铁电态在发生相变时,其热释电系数更高,发展前景广阔。电卡材料的综合性能指标可以用电卡系数ξmax来表示,其可以使用下列公式进行计算[26]:

式中:ΔTmax为最大改变温度;ΔEmax为最大温度改变时电场的改变。表II.列出了一些铁电与反铁电材料的电卡性能,从表中可以看出AFE材料普遍显示了较高的ΔT。
IV.钙钛矿金属氧化物
就功能电介质材料而言,固态晶体材料占了相当大的比例。而在固态晶体材料中,具有钙钛矿结构的金属氧化物有着显著的位置。因此,有必要简要的介绍钙钛矿结构特点,及其铁电与反铁电的结构特点,从而为材料的设计提供理论依据。现今已发现的铁电体相当一部分都是具有ABO3型的钙钛矿结构(如图13所示)的金属氧化物。钙钛矿金属氧化物的稳定性广泛使用Goldschimidt给出的容忍因子(tolerancefactor(t))表达式(公式12)来讨论[37]:

式中:RA为A位离子的半径;RB为B位离子的半径;RO为氧离子的半径。然而,在一些晶体中,A、B位离子与O离子之间的相互作用,并非完全以纯离子键的形式结合。如果它们之间存在电子轨道杂化的作用,而导致部分的共价键属性,就会影响离子之间的实际距离。因此,(公式12)中的A、B位离子与O离子的半径之和,则被修订为他们之间的键长[38],即:

式中:RA-O为 A–O键长;RB-O为 B–O键长。研究结果表明:当t约在0.8~1.05范围内时,分子通式为ABO3型的金属氧化物可以保持钙钛矿结构[39]。

表II.当前开发的一些FE与AFE材料的电卡性能

图13.立方钙钛矿金属氧化物的晶体结构(以A位阳离子为中心)
A.铁电扭曲
研究者们发现[40]:“容忍因子反映的实际上是钙钛矿晶体中离子的密排性质。”在 ABO3型钙钛矿结构的矩阵中,离子的密排方向有两个(如图14所示):一个是“-A-O-A-”离子线性链所在的方向(即:<110>晶向族);一是“-B-O-B-”离子线性链所在的方向(即:<001>晶向族)。当t=1时,在这两种线性链上都保持相似或相近的离子堆积密度。因而,立方钙钛矿稳定结构存在如BaZrO3和SrTiO3。当t>1时,立方晶胞在“-A-O-A-”离子链所在方向上紧密排列,而在“-B-O-B-” 链上的并非紧密排列,具有较大的间隙。B位离子受到周围氧离子的长程库仑力的影响,在氧八面体中发生自发极化(如图 15所示),从而呈现出铁电扭曲如: BaTiO3和 KNbO3等。而它们的自发极化强度都来自 B位阳离子自发极化的贡献。而自发极化的大小则与B位离子的极化率、电子结构、轨道杂化等因素有着密切关系。而当容忍因子 t<1时,立方晶胞在“-B-O-B-”离子线性链上密排。而“-A-O-A-”离子线性链所在方向上并非紧密排列。此时,氧八面体中的B位离子收到短程排斥力的影响,而阻碍B位离子的自发极化。而铁电扭曲主要来自于A位离子的自发极化。但实际情况是,在不考虑其他因素诸如电子轨道杂化(具有独特的 6s2孤电子对的Bi和Pb元素)等情况下[41],相当多的t<1的ABO3型钙钛矿金属氧化物并不具铁电性。

图14.钙钛矿原子结构密排方向示意图
B.反铁扭曲
标准的ABO3型立方钙钛矿结构,A位的配位数:CN=12,而B位的配位数:CN=6。而配位数取决于A、B位离子与O离子的半径之比(r+/r-)[42]。
已知发现的绝大多数 ABO3型钙钛矿结构的氧化物,其 B位的离子半径在 0.55˚A~0.75˚A之间。除太小的外,其它离子的配位数CN≊6。成键的B-O八面体骨架得以维持。而A位离子的半径除过Ba2+、K+与O的离子半径相似并维持12的配位环境以外,其余离子半径都比O离子半径小。从而使得A位的离子不能维持稳定的12配位。为了维持钙钛矿结构,A位的配位数必须降到一个合理的范围,以达到其最稳定的状态。从而,B-O八面体发生扭转或倾斜(如图16所示),使得A位离子并没有与所有的O离子成键。一般t值越小,其氧八面体越趋于不稳定。

图15.立方钙钛矿结构中的B位离子发生自发极化而成铁电结构示意图

表III.阴阳离子半径比与配位数的关系
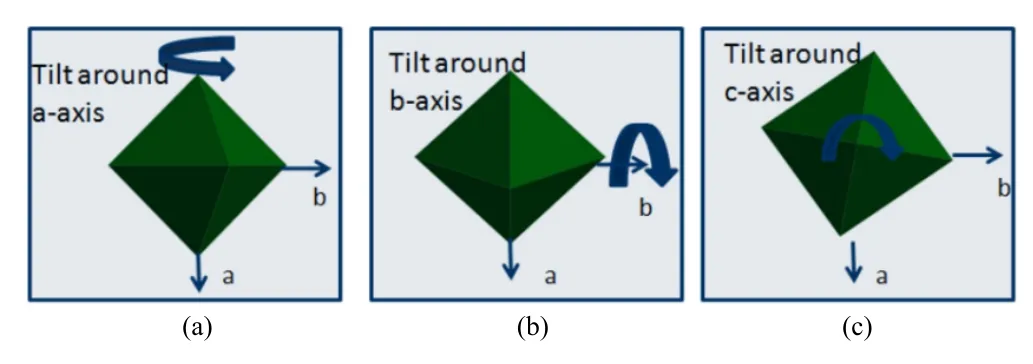
图16.氧八面体扭转示意图(a)围绕钙钛矿a轴,(b)围绕b轴,(c)围绕c轴[43]
而要保持有序的周期变化的晶体矩阵结构,相邻的两个氧八面体就要发生有规律的扭转,而钙钛矿结构的基本单元也相应的发生倍增,以形成超晶(如图 17所示)。英国晶体学家 Glazer教授发展并命名了独特的氧八面体扭转系统 (Octahedron rotation/Octahedron tilting),并预言了与一些空间群结构所适应的氧八面体扭转系统(如:氧八面体扭转系统 (a0,a0,c+),适合C4/mmb空间群)[44,45]。在氧八面体扭转系统中,符号“0”代表了氧八面体没有扭转。符号“+”代表了相邻两个氧八面体同相位扭转(In-phase tilting)。而符号“-”则意味着邻两个氧八面体以彼此相反的相位扭转(Anti-phase tilting)(如图18所示)。

图17.立方钙钛矿结构中的B-O八面体发生扭转而形成超晶示意图(中心圆球为A位阳离子)

图 18.相邻两层氧八面体扭转的二维示意图 (a)同相位扭转(In-phase tilting)(b)反相位扭转(Anti-phase tilting)[43]
而具有氧八面体扭转系统的钙钛矿超晶金属氧化物,其容忍因子t一般都小于1。它们最稳定的结构大多属于具有中心对称的正交晶系(如 GdFeO3、DyScO3、SrRuO3和 CaTiO3等),它们的晶体结构均为正交的空间群:“Pnma(a-,b+,a-)或者Pbnm(a-,a-,c+)”[46-48]。这些结构,都具有 A位离子发生反平行位移的特点(如图19所示)。但是,都不具备铁电性。因此,“八面体扭转”也可称之为“反铁扭曲 (Antiferrodistortive,AFD)”。最近,Benedek等人[38]认为:“在这些结构中,伴随氧八面体旋转的A位离子的反极化位移而非氧八面体旋转本身压制了铁电性的出现。”然而,这类ABO3型金属氧化物,至今依然未观察到双电滞回线并且在熔点附近,依然没有观察到介电异常的出现。

图19.SrSnO3的结构模型正显示A位 Sr离子的层状反向位移,而宏观不显示净极化[49]
V.反铁电的钙钛矿氧化物及其研究进展
当前,人们所普遍认为的具有反铁电性质的钙钛矿金属氧化物有:PbZrO3、PbHfO3、NaNbO3以及AgNbO3等。然而,它们晶体结构和电学性质却有着各自不同的特点。随着科学技术的不断发展、研究方法的日新月异,人们对这些化合物的理解也在不断的深入,从而一层层的神秘面纱被揭开并逐渐抵达真理的终点。
A.锆酸铅与铪酸铅
锆酸铅(PbZrO3)是目前广泛研究反铁电性起源的原型化合物。起初,在基于观察到的与BaTiO3相似的介电行为(如图20所示)上,PbZrO3被认为是铁电体,它的晶体结构属于四方晶系[50]。

图20.PbZrO3陶瓷的介电常数随温度变化的关系[51]
1951年,C.Kittle在基于立方钙钛矿结构基础上,提出“反铁电”概念及其理论的同时,Sawaguchi等人通过分析PbZrO3单晶样品的X-射线粉末衍射(X-ray di ff raction,简称XRD)图谱以及偏光显微照片认为其实际上属于正交晶系的超晶,并提出在a-b平面存在反铁电性(如图21所示)以及在c方向可能存在非常弱的铁电极化[52]。

图21.PbZrO3晶格的a-b平面显示了反向位移的Pb原子沿着赝立方钙钛矿结构的[110]方向,即正交晶系的a轴[52]

图22.PbZrO3晶体的电场–温度(E-T)相图(立方晶胞中的箭头,体现了晶体的极化。OR I:AFE-Pba2相;OR II:FE-C2mm 相;R I:FE-R3m 相;R II:FE-R3c相; OR I→OR II(E′c,T′c);OR II→RI(E′′c,T′′c);R I→R II(E′′′c,T′′′c);OR I→R I(Ec,Tc);OR II→R II(E′′′′c,T′′′′c))[60,61]
同年,美国学者Roberts对极化后的PbZrO3块材样品,进行压电测试,发现了可重复的非常弱的压电信号(d33=0.1 pC/N)[53]。50年代末,Jona等人[54]首次通过 XRD和中子衍射(Neutron di ff raction,简?称ND)实验确认PbZrO3的晶格结构属于具有非中心对称兼具极性的Pba2空间群,从而合理的解释了观察到的非常弱的压电效应。然而,进入80年代,Tanaka等人[55]则对PbZrO3晶体进行了电子衍射(Electron di ff raction,简称 ED)和汇聚束电子衍射(Converge beam electron di ff raction,简称CBED)实验,其结果则确认了该化合物属于中心对称的正交Pbam空间群。随后,Glazer以及他的学生[56]重新分析了ND数据均得到与Tanaka等人一样的结果,并宣称Roberts观察到的压电效应并非晶格本身的贡献,而与其他因素有关(如,热充电诱导的驻极体态)。除过它的室温晶体学结构,60年代末,几个研究组(Scott、Tennery以及Goulpeau等人)都认为PbZrO3在接近居里温度大概10~25°C的区域存在着具有属于三方晶系(R)的FE结构[57-59]。在随后的70年代末,基于高质量的PbZrO3单晶样品,Fesenko等人[60]系统的研究了该化合物的相变行为,并绘制出温度–电场(E-T)相图(如图22所示)。从相图上,确认了室温下三个电场诱导的相变:

并观察一个独特的六电滞回线。而陶瓷样品,由于击穿场强限制,到目前为止并未观 察到双电滞回线。Zhai等人在 (001)取向的 900 nm尺度的厚膜中在~60 0kV/cm的电场下,观察到双电滞回线,相变场强EF≈450 kV/cm[62]。Ayyub等人在 Si衬底上生长的PbZrO3薄膜中观察到,当厚度降低到~400 nm时,PbZrO3显示了铁电性[63]。最近,Mani等人通过第一性原理预测了PbZrO3能显示反铁电性的关键厚度则必须在5 nm以上[64]。目前,引起AFE相变的机制、及其反铁电性起源问题依然是凝聚态物理学、晶格动力学等领域的前沿热点。
对铪酸铅 (PbHfO3)的研究文献则相对比较少。Zr4+(0.72 ˚A) 的半径与 Hf4+(0.71 ˚A)的半径相当,与 PbZrO3相比较,PbHfO3显示了相似的晶体结构与介电行为。只不过,一个预示着相变的介电异常行为出现在约 160~180°C左右[65](如图 23所示)。随后,ND实验结果证实了PbHfO3具有与PbZrO3相同的晶格结构,同属正交晶系(O)的Pbam空间群[66]。而中间温度区域(额外的介电异常所在的温区范围),他们的晶体结构依然存在争议。Dernier等人[67]通过 XRD实验结果认为该化合物经历了 AFE(O)→FE(R)→PE(C)的相变变序列。最近,Kupriyanov等人[68]则通过XRD数据分析结果认为AFEO相经历了两个中间相 FEO(C2mm)和 FET(P4mm)。目前关于该化合物的E-T相图尚未报道。

图23.PbHfO3陶瓷的介电常数随温度变化关系曲线[65]
B.铌酸钠
铌酸钠 (NaNbO3)是一类具有复杂结构特征的 ABO3型金属氧化物。Shiratori等人[69]通过拉曼(Raman)图谱和XRD实验,研究了不同尺度晶粒的NaNbO3颗粒在室温下的晶格结构。结果发现,这些晶体都是具有氧八面体扭转的超晶。纳米尺度(70 nm)为 PE的Pmma相;亚微米尺度 (580~180 nm)的晶体则为 FE的Pmc21相;微米级 (1µm)也就是传统的固相法高温烧结的陶瓷粉体则为AFE的Pbcm相。而一般传统固相法高温合成的立方钙钛矿结构(C)的NaNbO3陶瓷样品,在降温的过程中经历了

6个多晶型相变而到达其最稳定的N相[56]。同时,在介电常数与温度变化的关系上(如图24所示),也披露了一系列介电行为的异常[70]。其中,最大介电异常,研究者们普遍认为是P↔R相变所致。几个高温多晶型相C、T2以及T1相都逐渐的通过XRD、ND以及XRD同步辐射等实验确定。它们分别对应于立方晶系的Pm¯3m相、四方晶系的P4/mbm相以及正交晶系的Cmcm(或Ccmm)相[71-74]。而S与R相,都显示了比较复杂的特征包括晶胞维度和空间群。Ahtee等人[72]以及Sakowski-Cowley等人[75]分别通过分析原位XRD实验数据认为:“S和R相是具有不同晶胞维度的正交Pnmm相。”

图24.NaNbO3陶瓷的介电常数、介电损耗随温度的变化曲线[56]
然而,Mishra等人[74]通过ND分析结果则宣称S和R相为具有不同的晶胞维度,但同属于正交Pbnm相。研究者们对N相的结构到目前为止,普遍没有争议,它的晶体结构为FE的R3c(FER)相,而P相则存在较大的争议。在早期的研究中,Yuzyuk等人[76]通过分析原位 Raman光谱和同步XRD辐射实验结果,则认为:“P相进一步经历了两个多晶型转变,最终演化成N相 (即,FER):

而近些年来,大多数研究者的分析结果普遍认为P相为具有Pbcm空间群的AFEO相。特别的,Mishra等人[77]通过原位加热和冷却的中子衍射实验的结果,则建议低温的AFEO(Pbcm)↔FER(R3c)的相变,显示了显著的热滞后特征,使得两相可以共存在非常宽的温度范围(-258°C~7°C(加热);-193°C~-258°C(冷却))。最近,研究者们通过二次谐波测试(SHG)和电子衍射等表征技术,相继报道了室温下存在 FEO的P21ma相[78],有的则在陶瓷块材中观察到亚稳态的 FEO相与 AFEO相共存[79,80]。 尽管 NaNbO3被认为具有反铁电性质,但是陶瓷块材在90 kV/cm的电场下,却观察到典型的兼具高饱和极化特征的FE型的电滞回线,而双电滞回线的报道则出现在同样测试电场下的单晶样品中,并且电场方向垂直于晶体的c轴,该相变认为是电场诱导的AFEO→FEO相变[81]。即:晶胞中反平行排列的偶极子在电场下呈现同向排列(相变前后宏观偶极子排列方向没有改变,都为[110]pc方向)。2015年,Shimizu等人[82]和Guo等人[83]分别采用具有更低钙钛矿容忍因子t的金属氧化物部分替代NaNbO3而形成NaNbO3基固溶体的办法来提高AFEO相与电场诱导的FEO相的自由能势垒。他们在 NaNbO3-CaZrO3和 NaNbO3-SrZrO3的固溶体陶瓷样品中,成功观察到具有AFE特征的双电滞回线(如图25所示),从而证实了这种策略对于稳定反铁电性的有效。

图 25. (a)NaNbO3-CaZrO3(CZNN)陶瓷的电滞回线(CZNN0:NN;CZNN2:0.98NN-0.02CZ;CZNN4:0.96NN-0.04CZ;CZNN5:0.95NN-0.05CZ)[82](b)NaNbO3-SrZrO3(CZNN)陶瓷的电滞回线(SZNN0:NN;SZNN4:0.96NN-0.04SZ;SZNN6:0.94NN-0.06SZ)[83]
C.铌酸银
铌酸银 (AgNbO3)与 NaNbO3的情况相似,AgNbO3也是一类具有复杂结构特征的金属氧化物。只不过,对于该化合物的研究文献与NaNbO3相比,相当稀缺。主要原因在于原材料–Ag2O高温容易被还原成Ag单质和O2,从而使得高质量的单相AgNbO3化合物比较难以合成[84]。1958年,两位学者几乎同时分别成功制备并报道了AgNbO3经历了O(正交)→T(四方)→C(立方)的相变序列,并推测其室温下可能具有反铁电结构[85,86]。80年代,Kania等人对 AgNbO3单晶和陶瓷样品分别进行了细致的研究[87,88]。他们基于赝立方 (Pseudo-cubic)钙钛矿结构基元,详细分析了XRD实验数据,并认为铌酸银氧化物经历了多个相变序列[87]:

细节的介电谱分析又建议有一个命名的O1相存在(如图26所示)[89]。但是差式扫描量热(简称DSC)的分析结果显示,在M1↔M2,M2↔M3以及M3↔O1相变处并没有存在吸热或者放热现象。随后的电滞回线、压电以及热释电的测试结果证实了M1相有极其微弱的铁电性 (PS=0.04µC/cm2,d33=0.24 pC/N,pt=-0.1 nC/cm2K)[90]。接着他们继续研究了Raman散射谱,并建议M2↔M3相变与Nb5+离子的可位移行为有着密切的关系[91]。Pawelczyk等人[92]和Petzelt等人[93]分别通过分析高分辨XRD实验数据认为M1和M2与铌酸钠的结构相同,为Pbcm相;M3为Cmcm或者Pbcn相;O相与T相分别为Pmnm相与P4/mbm相;而C相则为熟知的Pm¯3m相。2004年,Sciau等人[94]对陶瓷样品通过分析XRD和ND实验数据则认为三个M相均为Pbcm相,O相则为Cmcm相。T和C相与先前文献报道的结果一致。2007年,Fu等人[95]首次通过对陶瓷样品进行电学性质表征在 220 kV/cm的高电场强度下观察到类似反铁电(AFE-like)的双电滞回线,并且其最大应变量Smax=0.15%(如图 27所示)。然而,Sakurai等人[96]在SrTiO3衬底上生长的AgNbO3薄膜在~400 kV/cm的电场下却显示具有高极化的FE电滞回线,而双电滞回线只有在 (001)方向生长的铌酸银薄膜中观察到。最近,Ahn等人[97]在玻璃衬底上生长的AgNbO3薄膜得到与Sakurai等人相似的结果。

图26.AgNbO3陶瓷的介电常数、介电损耗随温度的变化关系[70]
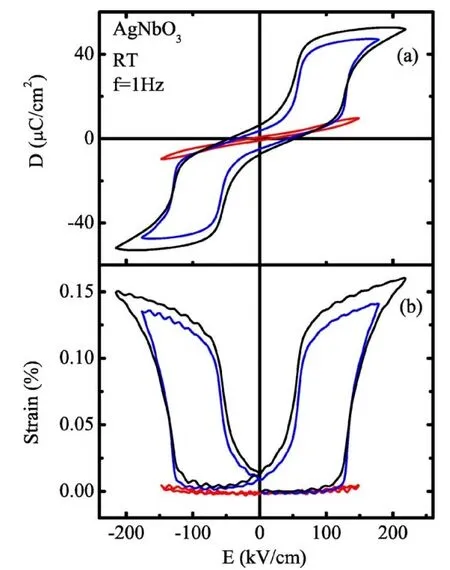
图27.AgNbO3陶瓷的(a)电滞回线(b)电致应变[95]
2009年,Levin等人[98]重新研究了铌酸银的微观结构。通过综合分析XRD、ND、ED以及EXAFS(扩展X射线吸收精细结构谱)实验数据认为:“在宏观尺度上,三个M相都显示了平均的Pbcm结构。”他们还指出几个介电异常则来源于亚晶格中Nb5+随机自发极化的沿着8个<111>pc可位移方向,在降温过程可自发位移的方向渐进式的减少并逐渐呈类似反极化(antipolar-like)的有序排列。而被认为是M2↔M3相变的扩散介电响应则来自于亚晶格中 Nb5+的反极化有序 (antipolar-like)↔无序 (random-polar)型转变。最近,Yashima等人[99]综合分析了铌酸银室温下的 XRD同步辐射、ND、ED以及 CBED图谱则宣称 AgNbO3室温下为具有非中心对称兼具极性的正交Pmc21相(如图 28所示),并具有亚铁电性(ferrielectric,简称 FIE)。

图28.AgNbO3的晶胞示意图(a)极性结构Pmc21(b)非极性结构Pbcm,箭头体现了Ag+与Nb5+离子的位移方向,绿色的多面体体现了Nb-O八面体[99]。
该晶体结构模型合理的解释M1出现的铁电性以及观察到的类似反铁电(AFE-like)双电滞回线。他们还认为的介电异常为Pmc21↔Pbcm相变所致。Niranjan等人[100]通过第一性原理计算发现这两种结构的自由能很接近并认为这两种相可以共存。同时,Moriwake等人[101,102]也通过第一性原理计算结果发现铌酸银的这两种结构转变在热动力学上是不可能发生的,并通过计算点缺陷形成能认为大量的Ag-O缺陷偶极子团簇应该存在Pbcm的晶格基体中。它们导致了观察到的AFE-like双电滞回线。而笔者通过分析XRD,SAED以及SHG实验数据和电性能表征发现AgNbO3陶瓷的基体结构更倾向于Pbcm空间群,但是局域存在结构为Pmc21的极性微区。同时,电性能表征发现有两种极性微区存在。其中,一种极性微区是不太稳定的,电场可以扩展其极性[103]。到目前为止,铌酸银的晶格结构与电性能之间的关联性依然比较缺乏统一的理解,有待未来进一步澄清和解决。
VI.具有反铁电扭曲的钙钛矿固溶体氧化物及其研究进展
尽管自然界中,具有反铁电性质的 ABO3型金属氧化物非常稀缺,研究者们发现对具有氧八面体扭转兼具铁电 (BiFeO3、(Bi1/2Na1/2)TiO3)或先兆铁电 (SrTiO3)性质的 ABO3型金属氧化物通过引入其他组元以形成固溶体的方法,依然可以创造出具有反铁电扭曲的化合物如 (Sr,Ca)TiO3(SCT)、(Bi,M=稀土元素)FeO3(RBF)以及 (Bi1/2Na1/2)TiO3-BaTiO3(BNT-BT)。
A.钛酸锶钙
钛酸锶钙((Sr,Ca)TiO3)是SrTiO3与CaTiO3的固溶体。SrTiO3(t≈1)和 CaTiO3(t≈0.964)都是具有先兆铁电 (incipient ferroelectric)或量子顺电(quantum paraelectric)特征的化合物。这类化合物室温并不表现出铁电性并且在温度降到极低时,依然没有铁电相出现。而介电常数单调增加并达到一个很大的饱和值(如图29所示)。

图29.SrTiO3、KTaO3以及CaTiO3陶瓷样品的约化介电常数ε(T)/ε(RT)与温度变化的关系,ε(RT)为室温下的介电常数 [104]。
目前,研究者们认为该现象是由于零点波动引起的量子起伏效应抑制了铁电有序而使顺电相保持稳定的结果。一般,量子起伏只对由质量轻的原子如氢、氦组成的化合物的结构和热力学性质有显著的影响,对于由重原子组成的化合物的结构和热力学性质并没有多少影响。然而,对于立方钙钛矿结构的化合物来说,由于他们的几种不同的晶体结构之间只有很小的自由能差别,因此量子起伏仍然对他们有着显著的影响从而保持一种平衡状态。电场、应力、杂质离子以及氧同位素等外界因素,都会打破这种平衡而诱导铁电相[104]。就晶体结构而言,SrTiO3室温下显示了立方Pm¯3m相而降温到~-168°C时发生了反铁扭曲转变而形成非极性的四方I4/mcm超晶相。该相一直保持到接近绝对零度 (~-273°C)[105]。
通过降低容忍因子t的策略,例如:离子半径更小的 Ca2+,部分的取代 Sr2+。研究者们发现,在 (Sr1-xCax)TiO3(SCT)固溶体中,随着 Ca2+含量的增加,一系列的相变行为被观察到。Bednorz和 Muller观察到当x=0.002时,在~-263°C观察到尖锐介电峰并几乎线性的向高温转移。随着Ca2+含量继续增加,当0.018 Ranjan等人[108]认为固溶体成分在此范围内的介电现象,是由于 AFE与 FE互竞争作用的结果。Mishra和Pandey分析了x=0.02和x=0.04两个固溶体成分的低温XRD实验数据。结果发现,SCT经历了从立方Pm¯3m相→四方非极性I4/mcm相→正交极性的Ic2m相的行为,表明出现了铁电有序[109]。当固溶体成分在0.12 对具有反铁扭曲(Antiferrodistortive, 简称AFD)的先兆铁电特征的钙钛矿化合物可以通过A位取代,进而降低钙钛矿容忍因子t而形成固溶体的思路,创造出具有反铁电的固溶体化合物。该原则同样也适用于具有AFD的铁电体。铁酸铋(BiFeO3,简称BFO)的容忍因子t≈0.96,室温下BFO的为三方晶系的R3c相并具有非常高TC。它的晶胞是由两个赝立方(Pseudo-cubic,简称PC)基元所组成,并且相邻的Fe-O八面体以反相旋转的方式围绕着赝立方 [111]pc晶轴排列,其巨大的饱和自发极化(Ps≈100µC/cm2)主要来源于 Bi3+和 Fe3+阳离子有序位移沿着 [111]pc晶轴[110]。不同于先前介绍AFD通常压制铁电性。研究者们普遍认为 BFO中的 Bi原子独特的“6s2孤电子对”与 O原子的 2p电子轨道之间的杂化扮演着重要的角色,而杂化作用影响到Fe2+的配位环境,最终有利于最低能量态的FER兼具(a-,a-,a-)型氧八面体扭转的R3c相的出现。由于BFO在650°C便开始有二次相产生而961°C便分解[111]。该特点阻碍了研究者们对其结构相变的深入理解。因此,它的PE相的结构目前存在较大的争议。Selbach等人[112]通过分析高温XRD图谱认为830°C左右发生了 FER3c↔PER¯3c转变。Palai等人[113]通过XRD、Raman以及DSC实验数据则认为陶瓷样品在TC以上存在两个PE相:正交相(TC~925°C)和立方相 (925°C~933°C)。Arnold等人[114]通过分析高温ND数据认为正交相的晶体结构为同样拥有AFD的Pbnm相。由于该化合物高的TC以及传统法制备的陶瓷样品具有高电导特点,从而影响了它的电学性能表征如:电滞回线。而对于该电学性质的表征主要在它的薄膜样品中。 具有钙钛矿结构的稀土铁酸盐(REFeO3(RE=稀土元素),简称RFO)化合物均显示了AFD的Pnma(或Pbnm)空间群结构并且容忍因子t随着稀土元素的原子序数的增加而逐渐的减小(如表4所示)[115]。如前所述,这些化合物的结构中均有A位阳离子发生反平行位移的特点,而宏观不显示净极化。 研究者们发现,当RFO与BFO形成固溶体化合物后,随着RE离子取代量的增加,TC逐渐降低,FE的R3c相逐渐转变为非极性的Pnma相,并在相变区域观察到AFE-like的双电滞回线和显著增强的压电活性。然而,对于相变区域的微观结构与电性能的关联性的理解,目前尚存争议。Rusakov等人[116]在 (Bi,La)FeO3陶瓷样品中,观察到在相变区域存在无共度相(incommensuate,简称INC)。Karimi等人[115]通过XRD、SAED、DSC以及Raman谱等表征与分析技术研究了不同 RE离子(RE=La,Nd,Sm,Gd)取代的BFO基固溶体的陶瓷样品,认为在相变区域存在具有类锆酸铅(PbZrO3-like)的结构隔离了R3c相与Pnma相,在温度-组分相图上,随着RE离子的半径减小该结构所存在的化学组成区域逐渐减小。随后,Levin等人[117]通过分析(Bi,Nd)FeO3固溶体陶瓷的XRD和ND数据认为该PbZrO3-like结构为AFE的正交Pnam相。Troyanchuk等人[118]通过分析 (Bi,RE)FeO3(RE=La、Pr、Nd、Sm、Eu、Gd、Tb以及Dy)XRD数据和电性能并绘制出了(Bi,RE)FeO3陶瓷固溶体中RE离子介电极化率与RE离子含量相图(如图31所示)。 图30.(a)Sr1-xCaxTiO3固溶体的介电常数随温度的变化曲线[106],(b)Sr1-xCaxTiO3固溶体的化学组成与温度的相图[107] 表IV.具有钙钛矿结构的稀土铁酸盐的容忍因子与其晶体学对称性[115] 图 31.RFO固溶体中 RE离子含量(RE element content)与离子介电极化率(Ion dielectric polarizability)的相图[118] 最近,Cheng等人[119]通过 HRTEM、SAED、STEM以及 EELS微观结构分析技术,研究了外延生长的 (Bi0.9Sm0.1FeO3)薄膜样品并观察到纳米级的反极化 (antipolar)团簇存在于 FER3c晶格矩阵中。他们认为观察到的具有 AFE行为的PbZrO3-like结构事实上归功于薄层状兼具交替极化的高密度铁电畴。同时,Kan等人[120,121]对 PLD法生长的 (Bi,RE)FeO3(RE=Sm、Gd以及Dy)薄膜样品通过2DXRD技术对相变区域的结构进行了表征也并未观察到可以定义的AFE相,而观察到具有1/4(011)超晶衍射斑点的局部正交结构(10~20 nm的有序尺度,并非AFE相)与占主导的FER3c共存,并认为这种相共存区域实质上归功于化学压力诱导的准同型相界(MPB),从而解释显著提高的电机械耦合性能(如图32所示)。该现象与在BFO陶瓷中等静压力诱导的R3c→Pbma(与 PbZrO3同结构)→Pnma的相变序列相似。 他们进一步通过第一性原理计算了BFO的R3c相与其他极性和非极性相的自由能差,并认为观察到的 AFE-like的双电滞回线(如图 33所示)并非典型的 AFE↔FE相所致,而是电场诱导的非极性的Pnma→FER3c相变。该事实再一次引发研究者们对AFE体定义的讨论。 图 32.(a)(Bi,RE=Dy、Gd、Sm)FeO3薄膜2D-XRD图谱中的1/4(0,1/4,7/4)和1/2(0,1/2,2)超晶斑点强度以及介电常数 (ε33)与 RE元素取代两的关系,1/4超晶斑点的出现与反平行位移的阳离子(AFE)有序相关;1/2超晶斑点来源于非极性Pnma结构;(b)温度–组分相图[120] 图33.(Bi,RE=Dy、Gd、Sm)FeO3中平均A位阳离子半径rave大小与电滞回线的关系[120] 钛酸铋钠(Bi1/2Na1/2)TiO3是目前研究比较广泛且争议很大的一类具有铁电性质的金属氧化物。该化合物A位被相同比例的Bi3+和 Na+离子所占据而保持整体上的A2+价,而Ti-O八面体则支撑了整个结构的框架,其容忍因子t≈0.945。而A位的Bi3+与Na+独特的化学环境,Bi和Ti原子各自的电子结构以及它们的壳层轨道电子(Bi:6s2、Ti:3d)与O(2p)壳层轨道电子之间的杂化作用等因素有利于阳离子发生不同程度的位移。在这些因素的共同协调下,导致了该化合物容易形成氧八面体扭转兼具阳离子位移的超晶结构并显示了独有的、显著区别与其他同样拥有Ti-O八面体的铁电氧化物(如:BaTiO3、PbTiO3)的电学性质(如图34所示,200°C左右出现的介电弛豫以及320°C出现的弥散介电峰等区别与其他典型铁电体的介电响应)。 图34.钛酸铋钠 (BNT)陶瓷样品的介电常数随温度的变化曲线,实现为升温过程,虚线为降温过程[122] 在早期的研究中,Sakata等人[123]通过铁电测试确认了剩余极化在 200°C附近显著降低。Pronin等人[124]通过 DSC分析确定了 200°C和 520°C附近出现了显著的热异常现象表明了两种一级多晶型相变行为(即,α→β→γ)。并且前者显示了显著了热滞后(~60°C)特征。通过这些迹象,研究者们认为BNT在200°C~320°C温度范围,具有AFE性质。Zvirgzds等人[125]对BNT单晶样品分析了XRD数据,认为BNT经历了两个晶型相变。它们分别是立方→四方 (~520°C)和四方→三方 (~200°C)相变。三个相的空间群分别为:Pm¯3m、P4mm 以及R3m。 随后,Vakhrushev等人[126]通过分析原位中子衍射数据并基于观察到的象征氧八面体扭转的超晶衍射线则确定了BNT的室温晶体结构为三方R3c相。与BiFeO3相似,该结构是由两个毗邻的TiO6八面体沿着[111]pc方向以反相扭转(a-,a-,a-)的方式组成的超晶。它们进一步分析了变温的中子衍射以及中子散射数据观察到与R3c结构相关的超晶衍射面彻底消失在~320°C,而另一种象征氧八面体同相扭转的超晶衍射面则开始出现在~220°C。它的衍射强度在~320°C抵达最高值并在~520°C显著降低。因而,220~320°C的温度范围被考虑为三方与四方相的共存。这两种类型的超晶衍射被Soukhojak等人[127]通过分析电子衍射图谱进一步证实。 Jones和 Thomas[128,129]通过分析中子衍射与二次谐波(SHG)数据确定了四方相的空间群为具有氧八面体关于c轴()[001]pc方向)同相扭转(a0,a0,c+)兼具极性的P4bm相,并发现该结构中B位的Ti4+离子与A位的Na+与Bi3+的自发位移量沿着[001]方向呈现不相等的反平行排列而导致了结构中出现的非常弱的净极化特征(即,weakly polar),从而确定了BNT化合物的AFE-like性质(如图35所示)。 图35.BNT的四方P4bm结构示意图(a)晶格[001]方向的投影图: ω:氧八面体旋转角度;圆圈:Bi3+/Na+;(b)晶格 [010]方向投影图,黑色圆点:Ti4+;圆圈:Bi3+/Na+;箭头:位移方向[128] 近年来,Gorfman等人[130]和 Aksel等人[131]分别对 BNT单晶样品和退火的陶瓷粉体样品通过高分辨率单晶衍射和光学双折射显微镜以及同步辐射 XRD技术认为室温下的结构为具有更低对称性的单斜Cc(a-,a-,c-)相而非先前研究者们认为的R3c(a-,a-,a-)相。Rao等人[132,133]详细的分析了不同BNT样品的状况下(包括退火的陶瓷粉体、研磨的陶瓷粉体、不同电场极化下的陶瓷块体以及研磨和退火后的极化过的陶瓷的粉体)的XRD数据发现晶体结构对外界环境(如:热、应力以及电场)非常敏感并认为在均衡状态下,室温晶体结构为Cc与R3c共存,应力和电场刺激均导致Cc转变为R3c相。 然而,不同于平均晶体结构的分析手段(如:ND和 XRD),TEM 和 SAED技术则提供了另外一幅结构蓝图。Dorcet和 Trolliardy[134]通过高分辨率透射电镜 (HREM)微观形貌照片结合选区电子衍射 (SAED)技术研究了 BNT的室温晶体结构认为在R3c(a-,a-,a-)晶格矩阵中存在几个晶胞厚度的 (001)取向的四方扭曲 (a0,a0,c+)局域结构并建议该现象导致了观察到的 200°C以下出现的介电弛豫现象。该研究团队[135,136]又重新研究了 BNT的多晶型相变行为认为当温度超过 200°C,一个拥有 (a-,b+,c-)的调制非极性的正交Pnma相薄层逐渐从R3c晶格矩阵的微观孪晶界处形成并与之共生,并认为该特征引起了观察到的 AFE-like行为。在 300°C附近Pnma相彻底取代了R3c相并通过二级相变 (~320°C)的方式进展性的改变成为只具有(a0,a0,c+)的顺电四方P4/mbm相并保留了局部的极性四方的P42/mnm纳米微区。Beanland和Thomas[137]对BNT无缺陷的单晶样品通过先进的数字电子衍射技术 (包括SAED和CBED)确认了纳米尺度的R3c结构并通过传统的TEM技术在存在不同缺陷类型的区域观察到一些复杂的微观组织(包括反相边界、畴壁、高密度的R3c纳米尺度的孪晶以及四方局部结构),并认为他们的相互作用导致了平均结构分析手段得到的Cc相。基于宏观与微观分析手段得到的结果的矛盾,最近,Levin和Reaney[138]通过几种不同的TEM分析手段并结合电子衍射技术综合分析了BNT在不同尺度的下的微观结构焦点在氧八面体扭转体系而非某个具体的、特定的相去处理BNT的局域与平均结构的问题。他们提出了BNT的微观结构实际是由具有(a-,a-,c+)氧八面体扭转框架的纳米孪晶所组成。只不过,在孪晶内,同相旋转(a0,a0,c+)的氧八面体有序长度比反相旋转的(a-,a-,a0)的氧八面体有序长度要少一个数量级以上。与此同时,阳离子自发位移呈现部分有序的排列沿着[111]与[001]方向。该思路的提出,使得人们对不仅仅是BNT,可能对一大类具有八面体扭曲兼具阳离子位移的化合物的错综复杂的晶体结构的研究具有重大借鉴意义。 如前所述,平均与局部结构分析方法(ND、XRD以及ED)都认为BNT在200°C以上存在具有AFE-like行为的组织,人们利用通过添加第二组元与BNT形成固溶体的思路,通过组分调节,使得该组织可以转移到室温附近,从而开发出一系列以BNT为基础的固溶体化合物。这其中,具有代表性的固溶体化合物便是钛酸铋钠–钛酸钡((Bi0.5Na0.5)TiO3-BaTiO3,简称 BNT-BT)。同时,该固溶体化合物也是存在争议最大的固溶体化合物。争议主要是来自于研究者们对 BNT-BT化合物MPB区域的结构–性能关联性的认识。早在1991年,日本研究者[139]就成功制备了 BNT-BT二元多晶固溶体并研究了该固溶体陶瓷样品极化之后的晶体结构与电学性质,并绘制出一个建议的相图(如图 36所示),一个几乎垂直的铁电三方(FR)与铁电四方(FT)准同型相界(MPB)坐落在x=0.06~0.07范围内。 图36.1991年出版的经过极化之后的BNT-BT二元固溶体的温度–组成相图,P:PE相;AF:AFE相;Fα:铁电菱方相;Fβ:铁电四方相[123] 近十几年来,对于化学组成接近MPB及其内部的微观组织、结构细节以及电学响应的研究,取得了显著的进展,尽管依然伴随着激烈的学术争辩。采用平均结构的分析手段如:XRD技术。研究者们认为MPB附近组分未经极化的结构显示了平均的顺电立方钙钛矿结构,很多文献将其冠以赝立方(pseudo-cubic)的称呼,以待以后解决。早期的介电表征确认了 MPB组分显示了类似弛豫体的介电行为。后来,F.Craciun等人[140]通过研究极化前后 XRD与复介电极化率图谱的变化证实了 MPB附近组成具有与弛豫铁电体(PMN、PST)相似的介电行为,并认为随着组分的变化,存在电场诱导与自发的弛豫态-铁电态转变(如图37所示)。从而阐明MPB区域应当存在极性纳米微区 (PNRs)。 除此之外,另一个突出的电学性质,就是双电滞回线以及电场诱导的巨大的应变(Electric- field induced strain)响应(如图38所示)。出现的双电滞回线迹象确认了(部分)可逆的电场驱动的弛豫(Relaxors)-FE或AFE-FE态转变存在,高温为完全的不可逆。到底是哪种目前学术尚存争议。 Simons等人[142]采用高分辨率中子衍射的分析手段,研究了 MPB区域靠近FR相一侧组分(x=0.06)的赝立方结构与巨大的电致应变响应的相互关系并认为其结构为具有细微四方畸变兼具(a0,a0,c+)型氧八面体扭转与细微三方畸变兼具(a-,a-,a-)型氧八面体扭转共存。在电场的作用下,赝立方结构转变为FR(R3c)相。Daniels等人[143]通过高能X-ray射线实验研究了MPB区域靠近FT相一侧组分(x=0.07)的电场诱导结构变化行为,发现了赝立方结构则转变为FT相。这些结果证实了介电行为的研究结果(即,Relaxors-FE态转变)。基于更先进的技术,Kitanaka等人[144]通过精修高能ND和高能同步X-ray辐射实验数据,认为x=0.07组分所谓的赝立方结构实际上为单相的具有AFE-like行为的P4bm极性结构,意味着BNT中的高温(Td以上)具有AFE-like行为的组织转移到室温附近。采用微区结构的分析手段, Ma和Tan[145]通过分析陶瓷样品的TEM和SAED图谱研究了未经极化的BNT-BT固溶体的多晶样品认为处于MPB区域靠近四方相一侧的化学组成的晶体结构为具有纳米畴结构的AFE-like四方P4bm相,并首次提出了“弛豫AFE相”的概念(如图39所示),而观察到的弛豫介电响应、双电滞回线与大应变响应与该结构的出现密切相关。 然而,Guo等人[141]分析的电子衍射结果却提供了另外一副图像去解释x=0.07组分的宏观电学性质。他们视乎并不太认同具有纳米畴结构的P4bm对称性的AFE-like相,而观察到的赝立方晶格矩阵中具有氧八面体扭转与阳离子可位移的无序行为导致了观察到的介电弛豫行为并与电场下观察到的双电滞回线与巨大的应变响应密切相关。由于在BNT中单斜Cc相的发现,Ma等人[147]通过TEM的分析手段,又提出了一个新单斜(a-,a-,c-)与三方的R3c(a-,a-,a-)的相界,出现在x=0.03。最近,Garg等人[148]发现与纯BNT类似,BNT-BT样品,特别是MPB附近的组成,其平均晶体结构不仅容易受电场的影响并且对外部机械应力也相当敏感,不同条件下处理的(如:研磨、退火)的陶瓷粉体的室温晶体结构都有很大的区别。 从钙钛矿容忍因子的角度来说,笔者认为BNT的容忍因子t<1。而BT的容忍因子t>1。在BNTBT固溶体中,随着BT含量的增加,容忍因子t应当逐渐增加。MPB组分的固溶体的平均容忍因子t≈1。该特点与XRD观察到很难定义对称性的赝立方结构吻合。而随着BT含量升高x>0.1,固溶体结构转变为没有AFD畸变的四方P4mm相。MPB区域组分的微观组织的形成应当是AFD与FE畸变的相互竞争与作用的结果。该特征导致了电场诱导的双电滞与巨应变响应。 图37.0.94BNT-0.06BT陶瓷的介电常数实部ε′与虚部ε′′随温度的变化(Td:去极化温度;T2:三方–四方相变温度;TVF=冰冻温度)[140] 尽管,美国物理学家 C.Kittle通过“双子晶格”模型提出了反铁电的概念,并通过唯像理论,预言了反铁电体的存在。然而,关于反铁电性的物理机制依然是当前研究的热点。物理学家认为,反铁电与铁电相变的驱动力来自于高温参考相的第一布里渊区晶格振动模的“软化”。如前面所述,具有反铁电位移的钙钛矿氧化物有很多如GdFeO3、SrSnO3等。它们的晶体结构拥有非极性的Pnma(a-,b+,a-)或Pbnm(a-,a-,c+)结构。然而,这类化合物,至少在电学行为上,不能够被考虑为典型的反铁电体。研究者们认为,该畸变的空间群结构是由 AFD与 AFE模耦合产生[149,11]。其中,两种模分别位于区边界的 M和R点,它们分别产生了氧八面体围绕着[001]pc轴扭转(即,(a0,a0,c+))和氧八面体围绕着 [110]pc轴扭转即,(a-,a-,c0))。这两种非极性模相结合构成了(a-,a-,c+)的氧八面体扭转系统。与此同时,位于X点的AFE模也是对称性允许的,该模产生了A位阳离子的反平行位移沿着[110]pc方向。然而,该AFE模的参与并非驱动相变的最主要力量,它是次级的。而位于区边界的M与R点晶格模的不稳定是造成该类AFE结构产生的关键。 从材料物理性质的角度,人们普遍认识的具有反铁电性的钙钛矿氧化物,不仅体现在晶格结构上,还体现在电学行为上。如前面所介绍 PbZrO3、NaNbO3等。这类材料普遍存在着与之竞争的相近能态的极性相,如 PbZrO3居里温度附近发现的R3m相;NaNbO3与 AgNbO3室温下分别发现的P21ma与Pmc21相。甚至,有些材料的电学行为的考虑超越了AFE标准结构的限制,如Bi0.5Na0.5TiO3中发现的具有阳离子AFE位移兼具极性结构的P4bm相。可见在相变过程中,AFE与FE扭曲存在着竞争是这类材料中出现AFE特征的内部属性。 PbZrO3是研究这类材料中反铁电性起源的理想钙钛矿氧化物。L.Bellaiche教授课题组通过第一性原理计算[150],提出了三种晶格模(分别位于第一布里渊区的Σ、R和S点)的线性合作耦合机制,它们的耦合作用促成了AFE相变的发生。并强调:“在几种能量相近的非极性相与极性相中,AFD晶格模扮演着至关重要的角色,形成了最终的非极性兼具AFE位移的Pbma超晶相。”该机制与前面提到的具有AFE特征的Pnma结构类似,都强调AFD模在驱动AFE相变的重要性。然而,A.K.Tagantsev等人[151]则认为控制AFE-PE相变的三种模分别为位于Σ、R和Γ点。Σ点产生了A位Pb离子的AFE位移;R点产生了AFD扭曲;而Γ点位于区中心位置,是极性模。它产生了异常的介电响应。他们进一步了细节的分析了 PbZrO3的介电行为与非弹性 X射线散射、X射线漫散射以及布里渊光散射的实验数据。认为:“驱动PbZrO3中AFE相变的驱动力来自于铁电模的软化,通过应变梯度/柔性电的耦合作用,转变系统成为丢失了无共度(INC)的AFE态,而适应Pbma空间群结构。”早前人们在化学改性的 PbZrO3陶瓷中发现了四方的AFE态中出现的INC调制,可能是对该机制有力的支持[152]。可见,在具有介电异常的AFE结构中,AFD和 FE模的初始不稳定,应当扮演着同等重要的角色。缺一不可。另外,通过前面总结关于PbZrO3、NaNbO3等化合物尺寸效应研究结果,笔者发现,随着薄膜厚度或者晶粒尺寸的降低,它们都经历了从AFE相→FE相→PE相的变化。这个特点视乎也预示着铁电模是最先陷入不稳定的。 图 38.0.93Bi0.5Na0.5TiO3-0.07BaTiO3组成陶瓷样品不同温度下的电致应变曲线(a)bipolar应变曲线(b)unipolar应变曲线[141] 图39.未经极化的BNT-BT二元固溶体陶瓷的温度–组成相图[145,146] 在无铅材料中,AgNbO3的Pmc21相与 Bi0.5Na0.5TiO3(BNT)的P4bm相,这两种结构都是具有A、B位离子AFE位移特点。然而,他们的对称性却是极性空间群,暂且称之为AFE-like结构。一些文献也称该类结构为亚铁电FIE。然而,亚铁电结构是相当罕见的。人们只是在液晶材料中发现了它的存在。而目前,关于钙钛矿中出现的该类结构的起源的研究较少。只不过,在具有BO6八面体的层状(类)钙钛矿铁电化合物中(如异质结的双钙钛矿、Dion–Jacobson相、Ruddlesden–Popper以及Aurivillius相等)。这些铁电结构中,都具有阳离子反平行位移的特点。如图40显示了Ca3Mn2O7铁电结构中阳离子层状位移图[153]。该结构中,Ca离子与氧八面体中的Mn离子都显示了反平行位移特征,而宏观上保留了净的FE极化。Mulder和Benedek等人通过第一性原理[154,155],计算了超过 16种这些层状(类)钙钛矿氧化物的结构,提出了“三线耦合”机制。不同于人们所认识的铁电体(驱动相变的源动力来自于自发极化P或软化的区中心极性模),该机制认为:“驱动 FE相变的源动力来自于区边界的AFD模。两个AFD模首先陷入不稳定并相互结合,再与第三个极性模耦合,导致了极性结构的出现。因此,他们是杂化非正规(hybrid improper)的铁电体。”同时,该机制强调,极性模不是不稳定的,只是它的出现归功于与AFD模的耦合。然而,这类结构并不会发生电场诱导的 FE-FE相变行为。从这个特点考虑的话,AgNbO3与BNT中出现的AFE位移兼具极性的空间群结构或FIE特性,驱动相变的首要源动力应当来源于区中心而非区边界。 图 40.Ruddlesden–Popper相的 Ca3Mn2O7层状阳离子极化 为应对全球日益剧增的大气污染状况和人类健康所面临的潜在威胁,“环境友好”的无铅材料已然成为近些年来全球科学家与工程师们关注的焦点。近十几年来,研究者们在无铅压铁电领域已取得了瞩目的成就。然而反铁电、特别是无铅反铁电领域的发展却显得相对滞后。因此,开发和探索无铅反铁电材料具有广阔的前景。基于这样的大背景下,笔者介绍了反铁电的概念、定义、电学性质以及潜在的应用前景。从材料角度,笔者重点介绍了具有AFE的钙钛矿氧化物的结构特点并详细的总结了当前人们普遍所认为的具有AFE性质的钙钛矿氧化物和钙钛矿固溶体化合物的晶体结构与电学性质的研究进展情况,并就当前从晶格动力学角度,去阐述钙钛矿氧化物中出现的反铁电性起源问题的研究情况做了评论,期望对即将或已经从事AFE领域(可能不仅仅局限于“无铅”)的材料基础和应用研究的科研工作者加深对其的理解与认识。 1951年,美国物理学家 C.Kittle类比于FE体,基于钙钛矿的简单“双子晶格”的结构模型,提出“AFE”概念。基本思想是:“相邻子晶格中的自发极化呈现反平行排列,而整个晶体不显示净的极化”,并从唯像理论的角度,预言了该物理现象的存在。然而,笔者总结了在随后的几十年中,人们在对一些钙钛矿氧化物的晶格结构和电学性质的研究中却发现,从严格意义上讲,符合“AFE”晶体结构限制的化合物,却不存在人们所认识的反铁电体所具有的电学性质。然而,符合“AFE”电学性质的钙钛矿化合物,在晶体学结构上却普遍存在着“极性与非极性”的争议。另外,有些研究者也存在这样的观念:“AFE-FE态转变中,结构的极化方向没有改变 (也就是结构的对称性上也应当具有要求)”。这也是先前一些研究者们[120]认为:“稀土替代的铁电铋中观察到的双电滞回线来源于电场诱导的非极性的正交(Pnma)-铁电(R3c)相变而非AFE-FE转变。”的原因。在Pnma-R3c相变中,Pnma结构中的AFE位移是[110]pc方向,R3c的FE位移是在[111]pc方向。而第一性原理计算的FE位移在[110]pc的Pmc21极性相的自由能远大于极性R3c相,电场诱导Pnma-Pmc21转变需要的电场值为2000 kV/cm。而R3c相与Pnma相最接近。因而,认为该转变并非 AFE-FE转变。如果极化方向也作为电场诱导的AFE-FE转变标准的话,在铅基反铁电体中观察到的一些双电滞回线,也应当不能被考虑由典型的AFE-FE转变所致(如电场诱导的四方AFE(INC)-三方 FE(R3c)转变)。因此,当前人们所普遍认为的AFE以及固溶体化合物应当属于“广义的”。笔者认为:“高对称性的非极性相降温过程中转变为低对称性的具有AFE位移的非极性相,转变处伴随着大的介电响应,通过电场该非极性相可以转变为FE相。符合这样的条件才能是严格意义的铁电体。”通过总结人们所认识的具有反铁电性质的钙钛矿氧化物和固溶体化合物的研究情况,笔者发现通过元素取代,而降低母相化合物的钙钛矿容忍因子t的微观结构设计策略,是有一个有效的方法稳定反铁电性和创建具有AFE性质的固溶体化合物。减小容忍因子t可以提升氧八面体不稳定性。而氧八面体扭转,是钙钛矿基本单元倍增,构建超晶结构的关键所在。 通过总结前人从晶格动力学角度,对钙钛矿和类钙钛矿氧化物中出现的阳离子的AFE位移起源的研究结果来看,驱动具有“AFE”电学性质的AFE相变发生的动力来源依然尚未解决,但是至少AFE与FE存在竞争是这类AFE结构的内部属性。 人们对当前发现的反铁电材料的认识与理解尚存争议。这其中,不乏有些理解,是基于对反铁电体认识的不同。如何让不同的认识达成共识是眼下所面临的最大问题。我们不防用“广义”与“狭义”去看待他们。“广义”就是符合反铁电体的电学性质(微观结构上尚存争议)。“狭义”是即符合AFE结构特点又符合电学性质。诸如CaTiO3、SrSnO3等具有正交Pnma或者Pbnm结构兼具AFE位移的化合物应当不能被考虑为反铁电体。尽管,天然存在的反铁电材料是相当稀缺的,通过微观结构设计策略,依然可以创造出具有反铁电性质的化合物,更多的未知AFE固溶体化合物有待未来发现。当前对无铅反铁电材料的开发主要集中在Bi0.5Na0.5TiO3为基元的化合物。最近,Liu等人在BNT基化合物中报道了0.7%的应变响应(如图所示),尽管存在非常大的滞后。通过性能优化,在大位移致动器上显示出应用潜力。AgNbO3、NaNbO3是两个具有广阔发展前景的无铅AFE材料,特别是在陶瓷块体。 图 41.无铅材料中电场诱导的应变 (S)与大信号压电系数d33(pm/V)的关系[156] 如前所述,AgNbO3在低场下具有极其微弱的FE滞后,而高场确显示了双电滞回线,尽管存在弱的剩余极化。研究者们普遍认为 NaNbO3的 FE型电滞回线是电场诱导的不可逆的 AFE-FE相变所致。因而,该化合物可以被考虑应用于爆电换能电源的开发。而AgNbO3的结构与性能之间的相关性理解上,目前争议较大。从应用角度来说,由于AgNbO3室温下显示了双电滞回线特征,因而可以通过对其性能的调节与改善,在高功率、高密度介电储能电容器中有着潜在的价值。笔者之前对AgNbO3为基元的化合物的研究结果,清晰了显示了该化合物作为无铅储能电容材料的优势。当然,并不是说NaNbO3只能应用在爆电换能,而AgNbO3只能应用于高密度介电储能电容器。通过微观结构设计策略,AFE材料的所有应用它们都具有广阔的发展前景。当前,这两种无铅反铁电材料的掺杂改性、固溶体化合物的基础研究相对很少,需要研究者们在这两个材料进一步深入的探索。当然,稀土铁酸铋也是可以开发的AFE材料,但是由于该材料打开双电滞回线所需的电场超出了陶瓷材料的击穿电场范围(~300 kV/cm),因而薄膜材料中开发的价值优越于陶瓷材料。现如今,无铅反铁电材料的开发,还上升不到实际应用的层面。实验室探索与基础学术研究依然是未来主要的研究方向。当然,基于合成技术的革新,其他钙钛矿反铁电化合物的诞生,也不是没有可能。 致 谢 本文的完成特别感谢西安交通大学王大威教授、同济大学翟继卫教授和刘星博士给予的讨论与建议。感谢国家重点基础研究发展计划 (批准号:2015CB654602)、国际合作项目 (批准号:2013DFR50470,51761145024)和“111”计划 (批准号:B14040)给予的课题资助! 参考文献 [1]Martienssen W,Warlimont H.功能材料:磁性材料,电介质,铁电体和反铁电体[M].哈尔滨:哈尔滨工业出版社,2014 [2]R¨odel J,Webber K G,Dittmer R,et al.J.Euro.Ceram.Soc.,2015,35:1659-1681 [3]European Union,(February 13,2003).“Directive 2002/96/EC of the European Parliament and of the Council of 27 January 2003 on Waste of Electrical and Electronic Equipment,”Official Journal of the EuropeanUnion,pp.L37/24-L37/38,http://europa.eu.int/eurlex/pri/en/oj/dat/2003/l{\_}037/ [4]中国出台首部电子信息产品污染防治管理办法[J].化工科技市场,2005(3):58-58 [5]R¨odel J,Jo W,Seifert K T P,et al.J.Am.Ceram.Soc.,2009,92:1153-1177 [6]Kittel C.Phys.Rev.,1952,82:729-732 [7]基特尔.C.固体物理导论[M].北京:科学出版社,1979,73-76 [8]Lines M E,Glass A M.Principles and Applications of Ferroelectrics and Related Materials[M].Cambridge University Press,1977 [9]Mitsui T.“Ferroelectrics and Antiferroelectrics”in Springer Handbook of Condensed Matter and Materials Data[M],Springer online publishing group,2005,903-938 [10]Jona F,Shirane G.Ferroelectric Crystals[M].Pergamon Press,1962 [11]Rabe K M.“Antiferroelectricity in Oxides:a Reexamination”in Functional Metal Oxides:New Science and Novel Applications[M].Wiley online publish group,2013,221-244 [12]Hao X.J.Adv.Diectri.,2013,3:1330001 [13]Dang Z M,Yuan J K,Yao S H,et al.Adv.Mater.,2013,25:6334-6365 [14]Tian Y,Jin L,Zhang H,et al.J.Mater.Chem.A,2017,5:17525-17531 [15]Park S E,Pan M J,Markowski K,et al.J.Appl.Phys.1997,82:1798 [16]Berlincourt D.IEEE Trans.Son.Ultrason,1968,15:89-97 [17]吕可佳,李俊宝,邢建新,等.声学学报,2011,36:520-526 [18]Reynolds C E,Seay G E.J.Appl.Phys.,1961,32:1401-1402 [19]Jo H R,Lynch C S.J.Appl.Phys.,2014,116:074107 [20]Zhang Q F,Jiang S L,Zeng Y K,et al.J.Appl.Phys.2011,109:124111 [21]张清风.Pb(Zr,Sn,Ti)O3基反铁电陶瓷场致热释电效应与应用研究[D].武汉:华中科技大学,2012. [22]Yang T Q,Liu P,Xu Z,et al.Ferroelectrics,1999,230:181-6 [23]Sungtaek J Y.J.Electron.Pack.2010,132:041004 [24]Scott J F.Annu.Rev.Mater.Res.,2011,41:1-12 [25]Valant M.Prog.Mater.Sci.2012,57:980-1009 [26]Zhao Y,Hao X,Zhang Q.J.Mater.Chem.C,2015,3:1694-1699 [27]Mischenko A S,Zhang Q,Scott J F,et al.Science,2006,311:1270-1271 [28]Neese B,Chu B J,Lu S G,et al.Science,2008,321:821-823. [29]Peng B L,Fan H Q,Zhang Q,et al.Adv.Funct.Mater.,2013,23:2987-2992 [30]Saranya D,Parui J,Krupanidhi S B,et al.Ferroelectrics,2013,453:38-43 [31]Tuttle B A,Payne D A.Ferroelectrics,1981,37:603-606 [32]Thacher P D.J.Appl.Phys.,1968,39:1996-2002 [33]Qian X S,Ye H J,Zhang Y T,et al.Adv.Funct.Mater.,2014,24:1300-1305 [34]Wang JF,Yang TQ,Wei K,et al.Appl.Phys.Lett.,2013,102:152907 [35]Jiang X J,Luo L H,Wang B Y,et al.Ceram.Int.,2014,40:2627-2634 [36]Singh G,Bhaumik I,Ganesamoorthy S,et al.Appl.Phys.Lett.,2013,102:082902 [37]Li C,Soh K,Wu P.J.Alloy.Compd.,2004,372:40-48 [38]Benedek N A,Fennie C J.J.Phys.Chem.C,2013,117:13339-13349 [39]Randall C A,Bhalla A S,Shrout T R.J.Mater.Res.,1990,5:829-834 [40]施韬.钙钛矿型铁电材料设计思路及电子显微学研究[D].北京:清华大学,2016 [41]King G,Woodward P M.J.Mater.Chem.,2010,20:5785-5796 [42]胡赓祥,蔡珣.材料科学基础[M].上海:上海交通大学出版社,2000,31-36 [43]Johnson R.Phase Transitions in Ag(TaxNb1-x)O3thin films[D].United State of American,The Pennsylvania State University,2011 [44]Glazer A M.Acta Cryst.,1975.A31:756 [45]Glazer A M.Acta Cryst.,1972.B28:338 [46]Biegalski M D,Haeni J H,Trolier-McKinstr S,et al.J.Mater.Res.,2005,20:952-958 [47]Kennedy B J,Hunter B A,Hester J R.Phys.Rev.B,2002,65:224103 [48]Kennedy B J,Howard C J,Chakoumakos B C.J.Phys.Conden.Mat.,1999.11:1479-1488 [49]Mulder A T,Benedek N A,Rondinelli J M,et al.Adv.Funct.Mater.,2013,23:4810-4820 [50]Roberts S.J.Am.Ceram.Soc.1950,33:63 [51]Shirane G,Sawaguchi E,Takagi Y.Phys.Rev.,1951,84:476 [52]Sawaguchi E,Maniwa H,Hoshino S.Phys.Rev.,1951,83:1078 [53]Roberts S.Phys.Rev.,1951,83:1078 [54]Jona F,Shirane G,Mazzi F,et al.Phys.Rev.105:849-856 [55]Tanaka M,Saito R,Tsuzuki K.J.Phys.Soc.Jpn.,1982,51:2635-2640 [56]Corkker D L,Glazer A M,Dec J,et al.Acta Cryst.,1997,B53:135-142 [57]Scott B A,Burns G.J.Am Ceram.Soc.,1972,55:331-333 [58]Tennery V J.J.Am.Ceram.Soc.,1966,49:483-486 [59]Goulpeau L.Fiz.Tverd.Tela,1966,8:2469 [60]Fesenko O E,Kolesova R V,Sindeyev Y G.Ferroelectircs,1978,20:177-178 [61]Hao X,Zhai J,Kong L B,et al.Prog.Mater.Sci.,2014,63:1-57 [62]Hao X,Zhai J,Yao X.J.Appl.Phys.,2008,104:124101 [63]Ayyub P,Chattopadhyay S,Pinto R,et al.Phys.Rev.B,1998,57:R5559 [64]Mani BK,Chang C,Lisenkov S,et al.Phys.Rev.Lett.,2015,115:097601 [65]Shirane G,Pzpinsky R.Phys.Rev.,1953,91:812 [66]Corker D L,Glazer A M,Kaminsky W,et al.Acta Cryst.1998,B54:18-28 [67]Dernier P D,Remeika J P.Mater.Res.Bull.,1975,10:187-192 [68]Kupriyanov M F,Petrovich E V,Dutova E V,et al.Cryst.Rep.,2012,57:205-207 [69]Shiratori Y,Magrez A,J.Phys.Chem.B,2005,109:20122-20130 [70]Kania A,Kwapulinski J.J.Phys.:Condens.Matter,1999,11:8933-8946 [71]Glazer A M,Megaw H D.Phil.Mag.,25:1119-1135 [72]Ahtee M,Glazer A M,Megaw H D.Phil.Mag.,1972,26:995-1014 [73]Ishida K,Honjo G.J.Phys.Soc.Jpn.,1972,34:1279-1288 [74]Mishra S K,Mittal R,Pomjakushin V Y,et al.Phys.Rev.B,2011,83:134105 [75]Sakowski-Cowely A C,Lukazewicz K,Megaw H D.Acta Crystallogr.Sect.B,1969,25:851 [76]Yuzyuk YI,Simon P,Gagarina E,et al.J.Phys.:Condens.Matter.,2005,17:4977 [77]Mishra S K,Choudhury N,Chaplot S L,et al.Phys.Rev.B,2007,76:024110 [78]Johnston K E,Tang C C,Parker J E,et al.J.Am.Chem.Soc.,2010,132:8732-8746 [79]Guo H,Shimizu H,Mizuno Y,et al.J.Appl.Phys.,2015,118:054102 [80]Guo H,Shimizu H,Randall C A.Appl.Phys.Lett.,2015,107:112904 [81]Cross L E,Philos.Mag.,1955,46:453-466 [82]Shimizu H,Guo H,Reyes-Lillo S E,et al.Dalton Trans.,2015,44:10763 [83]Guo H,Shimizu H,Mizuno Y,et al.J.Appl.Phys.,2015,117:214103 [84]Valant M,Axelsson A,Alford N.J.Euro.Ceram.Soc.,2007,27:2549-2560 [85]Francombe M H,Lewis B.Acta Crystallogr.,1958,11:175 [86]Reisman A,Holtzberg F,J.Am.Chem.Soc.,1958,80:6503-6507 [89]Kania A.Ferroelectrics,1998,205:19-28 [91]Kania A,Roleder K,Kugel GE,et al.J.Phys.C:Solid State Phys.,1986,19:9-20 [92]Pawelczyk M.Phase Trans.,1987,8:273-292 [93]Petzelt J,Kamba S,Buixaderas E,et al.Ferroelectrics,1999,223:235 [94]Sciau P,Kania A,Dkhil B,et al.J.Phys.:Condens.Matter,2004,16:2795-2810 [95]Fu D,Endo M,Taniguchi H,et al.Appl.Phys.Lett.,2007,90:252907 [96]Sakurai H,Yamazoe S,Wada T.Appl.Phys.Lett.,2010,97:042901 [97]Ahn Y,Seo J,Son J Y.Appl.Surf.Sci.,2015,357:429-432 [98]Levin I,Krayzman V,Woicik J C,et al.Phys.Rev.B,2009,79:104113 [99]Yashima M,Matsuyama S,Sano R,et al.Chem.Mater.2011,23:1643-1645 [100]Niranjan M K,Asthana S.Soli.Stat.Commun.,2012,152:1707-1710 [101]Moriwake H,Fisher C A J,Kuwabara A,et al.Jpn.J.Appl.Phys.,2013,52:09KF08 [102]Moriwake H,Fisher C A J,Kuwabara A,et al.Jpn.J.Appl.Phys.,2012,51:09LE02 [103]Tian Y,Jin L,Zhang H,et al.J.Mater.Chem.A,2016,4:17279-17287 [104]袁敏,量子起伏对几种先兆性铁电体介电性质的影[D].山东:山东大学,2005 [105]Fujishita H,Shiozaki Y,Sawaguchi E.J.Phys.Soc.Jpn.,1979,46:581-586 [106]Bednorz J G,Muller K A.Phys.Rev.Lett.,1984,52:2289-2292 [107]Ranjan R,Pandey D.J.Phys.:Condens.Matter,2001,13:4251-4266 [108]Ranjan R,Pandey D,Sirugur V,et al.J.Phys.:Condens.Matter,1999,11:2233-2246 [109]Mishra S K,Pandey D.Appl.Phy.Lett.,2009,95:232910 [110]Zhao T,Scholl A,Zavaliche F,et al.Nat.Mater.,2006,5:823-829 [111]Catalan G,Scott J F.Adv.Mater.2009,21:2463-2485 [112]Selbach S M,Tybell T,Einarsrud M,et al.Adv.Mater.2008,20:3692-3696 [113]Palai R,Katiyar R S,Schmid H,et al.Phys.Rev.B,2008,77:014110 [114]Arnold D C,Knight K S,Morrison F D,et al.Phys.Rev.Lett.,2009,102:027602 [115]Karimi S,Reaney I M,Han Y,et al.J.Mater.Sci.,2009,44:5102-5112 [116]Rusakov D A,Abakumov A M,Yamaura K,et al.Chem.Mater.,2011,23:285-292 [117]Levin I,Karimi S,Provenzano V,et al.Phys.Rev.B,2010,81:020103(R) [118]Troyanchuk I O,Karpinsky D V,Bushinsky M V,et al.J.Am.Ceram.Soc.,2011,94:4502-4506 [119]Cheng C,Borisevich A Y,Kan D,et al.Chem.Mater.2010,22:2588-2596 [120]Kan D,P´alov´a L,Anbusathaiah V,et al.Adv.Funct.Mater.,2010,20,1108-1115 [121]Kan D,Long C J,Steinmetz C,et al.J.Mater.Res.,2012,27:2691-2704 [122]赵明磊,钟维烈,王春雷等人.钛酸铋钠系铁电体的相变研究[J].物理学报.2002,51:1856-1860 [123]Sakata K,Masuda Y.Ferroelectrics,1974,7:341-349 [124]Pronin I P,Syrnikov P P,Isupov V A,et al.Ferroelectrics,1980,25:395-397 [125]Zvirgzds J A,Kapostin P P,Zvirgzde J V,et al.Ferroelectrics,1982,40:15-11 [126]Vakhrushev S B,Isupov V A,Kvyatkovsky B E,et al.Ferroelectrics,1985,63:153-160 [127]Soukhojak A N,Wang H,Farrey G W,et al.J.Phys.Chem.Solids,2000,61:301-304 [128]Jones G O,Thomas P A.Acta Cryst.,2002,B58:168-178 [129]Jones G O,Thomas P A.Acta Cryst.,2000,B56:426-430 [130]Gorfman S,Thomas P A.J.Appl.Cryst.,2010,43:1409-1414 [131]Aksel E,Forrester J S,Jones J L,et al.Appl.Phys.Lett.,2011,98:152901 [132]Rao B N,Fitch A N,Ranjan R.Phys.Rev.B,2013,87:060102(R). [133]Rao B N,Ranjan R.Phys.Rev.B,2012,86:134103 [134]Dorcet V,Trolliard G.Acta Mater.,2008,56:1753-1761 [135]Dorcet V,Trolliard G.Chem.Mater.,2008,20:5061-5073 [136]Trolliard G,Dorcet V.Chem.Mater.,2008,20:5074-5082 [137]Beanland R,Thomas P A.Phys.Rev.B,2014,89:174102 [138]Levin I,Reaney IM.Adv.Funct.Mater.,2012,22:3445-3452 [139]Takenaka T,Maruyama K,Sakata K.Jpn.J.Appl.Phys.,1991,30:2236-2239 [140]Craciun F,Galassi C,Birjega R.J.Appl.Phys.,2012,112:124106 [141]Guo Y,Liu Y,Withers R L,et al.Chem.Mater.,2011,23 219-228 [142]Simons H,Daniels J E,Jo W,et al.Appl.Phys.Lett.,2011,98:082901 [143]Daniels J E,Jo W,R¨odel J,et al.Appl.Phys.Lett.,2009,95:032904 [144]Kitanaka Y,Ogino M,Hirano K,et al.Jpn.J.Appl.Phys.,2013,52:09KD01 [145]Ma C,Tan X.Solid State Commun.,2010,150:1497 [146]Ma C,Tan X.J.Am.Ceram.Soc.,2011,94,4040-4044 [147]Ma C,Guo H,Tan X.Adv.Funct.Mater.2013,23:5261-5266 [148]Garg R,Rao N B,Senyshyn A,et al.Phys.Rev.B,2013,88:014103 [149]BellaicheL,Iniguez J.Phys.Rev.B,2013,88:014104 [150]Iniguez J,Stengel M,Prosandeev S,et al.Phys.Rev.B,2014,90:220103(R) [151]Tagantsev A K,Vaideeswaran K,Vakhrushev S B,et al.Nat.Comm.,2013,4:2229 [152]Tan X,Ma C,Frederick J,et al.J.Am.Ceram.Soc.,2011,94:4091-4107 [153]Benedek N A,Mulder A T,Fennie C J.J.Solid StateChem.,2012,195:11-20 [154]Mulder A T,Benedek N A,Rondinelli J M,et al.Adv.Funct.Mater.,2013,23:4810-4820 [155]Benedek N A,Rondinelli J M,Djani H,et al.Dalton Trans.,2015,44:10543-10558 [156]Liu X,Tan X.Adv.Mater.,2016,28:574-578 Antiferroelectric materials exhibit potential applications in morden electronic technologies due to their phase transition behavior under electrical loading.Currently,“Environmental friendly”lead-free materials have been attracting more interest to global scientists and engineers due to the concern of serious air pollution and human health condition.However,the native antiferroelectric materials are very rare and the understanding regarding to their structure-property relationship yet remains to be a controversial issue in scienti fic community.Therefore,this review summarizes the progress of antiferroelectric perovskite oxides since the concept of“antiferroelectric” was introduced in 1951 by C.Kittle,and comments the origin of antiferroelectricity in terms of lattice dynamics based on the progress of researches on antiferroelectric perovskite oxides. Progess of Antiferroelectric Perovskite Oxides Tian Ye,Jin Li,Feng Yu-Jun,Zhuang Yong-Yong,Xu Zhuo,Wei Xiao-Yong Lead-free;Antiferroelectric;Pervoskite Structure date:2017-07-25 O412 A 10.13725/j.cnki.pip.2017.05.001 *E-mail:wdy@xjtu.edu.cn 1000-0542(2017)05-0155-27B.稀土离子取代的铁酸铋





C.钛酸铋钠和钛酸铋钠-钛酸钡固溶体




VII.钙钛矿金属氧化物中的反铁电性起源



VIII.全文总结与展望
A.全文总结
B.展望

Electronic Materials Research Laboratory,Key Laboratory of the Ministry of Education&International Center for Dielectric Research,Xi’an Jiaotong University,Xian 710049,China
猜你喜欢
无机材料学报(2022年6期)2022-08-25 12:26:28陶瓷学报(2021年3期)2021-07-22 01:05:06科学(2020年4期)2020-11-26 08:27:12新世纪智能(数学备考)(2019年9期)2019-10-16 11:44:58苏州科技大学学报(自然科学版)(2017年1期)2017-03-20 15:25:18物理学进展(2017年1期)2017-02-23 01:35:44湖北工业大学学报(2016年5期)2016-02-27 13:15:01云南师范大学学报(自然科学版)(2015年5期)2015-12-26 12:46:14大学化学(2015年5期)2015-09-18 08:43:48太阳能(2015年4期)2015-02-28 17:08:19
