
Alpers综合征2例报道并文献复习
2017-10-17 08:53:48史晓依张瑞杰
中风与神经疾病杂志 2017年9期
史晓依, 张瑞杰
Alpers综合征2例报道并文献复习
史晓依, 张瑞杰
Alpers综合征是一种常染色体隐性遗传的肝脑综合征,其典型的临床特征为难治性癫痫、应用丙戊酸后会发生急性肝衰竭、皮质盲和精神运动倒退。发病年龄为1个月~25岁,以婴幼儿常见,多在3岁内死亡[1]。Bernard Alpers首次于1931年报道了Alpers综合征的神经病理学及临床特征[2],此后对于本病的研究逐步深入,国内为北京大学第一医院包新华等首次于2008年通过基因突变分析确诊国内首例Alpers综合征[3]。尽管如此,由于本病发病率极低,国内相似文献报道并不多见。我院近期接诊2例此类患儿,特报道如下,同时对此类疾病特点进行文献复习。
1 临床资料
病例1:患儿,女,(31月龄)因“间断抽搐6 h”就诊于我院小儿ICU科,抽搐表现为右侧嘴角抽动,牙关紧闭,意识清楚,有自主呼吸,四肢无强直,面色正常,持续1~2 min可自行缓解,1~2 min后复发。1 h后患儿开始出现双眼向右上翻。2 h后开始出现右上肢僵直,双手握拳,双脚背屈,持续2~3 min后自行缓解,2~3 min后复发。3 h后开始出现头向右晃动,颈部僵直。查体:W:6.5 kg,H:75 cm营养不良貌,皮肤暗黄,双侧瞳孔对光反射略迟钝,咽充血,双肺呼吸音粗,可闻及大中水泡音,四肢肌张力低下。诊断为“癫痫,肺炎,运动发育迟缓,急性心肌损伤”,给予奥卡西平[10 mg/(kg·d)]等对症治疗无抽搐,好转出院,院外继续口服奥卡西平预防抽搐。11 m后(42月龄)因“间断抽搐11 m,痰鸣10 d,嗜睡2 d”就诊于我科,抽搐表现为眨眼、嘴角抽动,伴咀嚼样动作,逐渐出现手足抽动,持续约1 h。入院后给予抗感染、止抽等对症处理,结合基因检查结果,诊断“Alpers综合征、癫痫持续状态、肺炎、急性心肌损伤”,好转出院。院外间断抽搐发作,2 m后患儿去世。
既往史及家族史:G1P1,足月儿,因胎动减少选择性剖宫产,出生体重4.6 kg,脐带缠颈2 w。生后4 d因“进乳差,有呛咳”就诊于我院新生儿科,诊断为“巨大儿、新生儿高胆红素血症”,住院期间奶量为50 ml/q 3 h,自行进食良好。出院后进乳欲望差,奶量不能按计划增加。生后8 m就诊于我院行遗传代谢疾病检查未见异常,未予特殊处置。2岁时因“生长发育落后”就诊于长春市儿童医院及北京儿童医院,疑诊“脑瘫”,家长诉无特殊治疗。生后未定期监测体重及身长,1周岁时体重5~5.5 kg,2周岁时8.5 kg。生后6~8个月会翻身,10个月会坐,至今不会爬、站立及走路。具体开始说话时间不详,2周岁时会说4~5个单词,可持筷子及笔。否认癫痫家族史。
辅助检查:2次入院相关感染指标:WBC:17.38×109/L,N:0.88;PCT:0.95 ng/ml;WBC:19.13×109/L,N:0.78;CRP:99.60 mg/L;PCT:1.42 ng/ml。2次入院脑脊液检查:蛋白8.20 g/L,潘氏反应,白细胞16×106/L,红细胞3000×106/L,多核0.56,单核0.44。蛋白7.90 g/L,潘氏反应,白细胞8×106/L,多核0.25,单核0.75。2次均伴有心肌酶、转氨酶轻微升高。动态脑电图:异常脑电图,背景活动慢化,双侧后头部为主不对称,左侧波幅高且放电多,左侧后头部棘波、多棘慢波、慢波发放。(北京大学第一医院2016.01.11)基因分析报告:POLG基因有2个杂合变异,分别来自于父亲和母亲,(疾病/表型)为线粒体DNA缺失综合征-4型(Alpers型)。
病例2:患儿,男,11月龄发病,因抽搐先后4次入住我科。第1次(11月龄)因“持续左侧肢体抽搐4 h”入院,表现为左侧肢体抽动,手脚抽动明显,无颜面青紫,无意识丧失,每次持续约4~8 min可自行缓解,2~3 min后再发,诊断为“癫痫(局灶性发作、癫痫持续状态、持续性部分性发作)”,入院后咪达唑仑持续泵入下抽搐逐渐好转,后规律口服“奥卡西平[20 mg/(kg·d)]、托吡酯[5 mg/(kg·d)]”,病情控制尚可,左侧肢体未再抽搐,院外逐渐调整奥卡西平[50 mg/(kg·d)]、托吡酯[5 mg/(kg·d)]。第2次(13月龄)患儿无明显诱因出现右侧肢体抽搐,性质同前,自行给予水合氯醛灌肠后症状有所缓解,再次就诊我科,行基因检测结果回报诊断为“Alpers综合征,癫痫、部分性发作、代谢性”住院9 d好转出院。院外规律口服“奥卡西平[62.5 mg/(kg·d)]、托吡酯[6.25 mg/(kg·d)]”。第3次(14月龄)因抽搐入院,表现为意识丧失,口唇发绀,双眼凝视,左侧肢体抽动,持续不缓解。入院后继续上调奥卡西平及托吡酯剂量,同时加用左乙拉西坦[15 mg/(kg·d)]起控制抽搐。第4次(15月龄)因抽搐入院,表现同前。“奥卡西平[67.5 mg/(kg·d)]、托吡酯[8.75 mg/(kg·d)]、左乙拉西坦[35 mg/(kg·d)]”联合作用下仍有间断抽搐,患儿家属自行要求出院,1 m后患儿死亡。
既往史及家族史:出生史及生长发育史正常。患儿姐姐3年前因间断抽搐4 m去世。
辅助检查:第1次入院WBC:5.50×109/L,N:0.71,CRP:9.97 mg/L。脑脊液蛋白:0.47 g/L。4次入院均伴有心肌酶及肝酶升高。头MRI:(1)双侧脑室旁异常信号,不除外髓鞘发育尚不完全所致;双侧脑室后角形态可疑改变。(2)双侧额部蛛网膜下腔略增宽。前3次EEG:弥漫性δ波为主慢波活动,前头部及后头部著,右侧Rolandic区慢波、或夹杂(多)棘波尖波,或(多)棘慢波接近持续发放;背景活动慢化(2.5~3 Hz中波幅δ波);弥漫性δ波为主慢波活动持续发放,右侧后头部棘慢波发放。基因检测报告回报(康旭医学检验所2016年5月24日):在受检者POLG基因发现复合杂合核苷酸变异,分别遗传自父母,其父母均只携带其中一个杂合变异。
病例特点:婴幼儿期发病,起病急,病情反复,呈加重趋势。以癫痫为首发症状,合并呼吸系统感染,伴有心肌、肝功损伤,脑电图提示背景活动慢化,经基因诊断明确为本病。最终死于癫痫持续状态(见表1)。
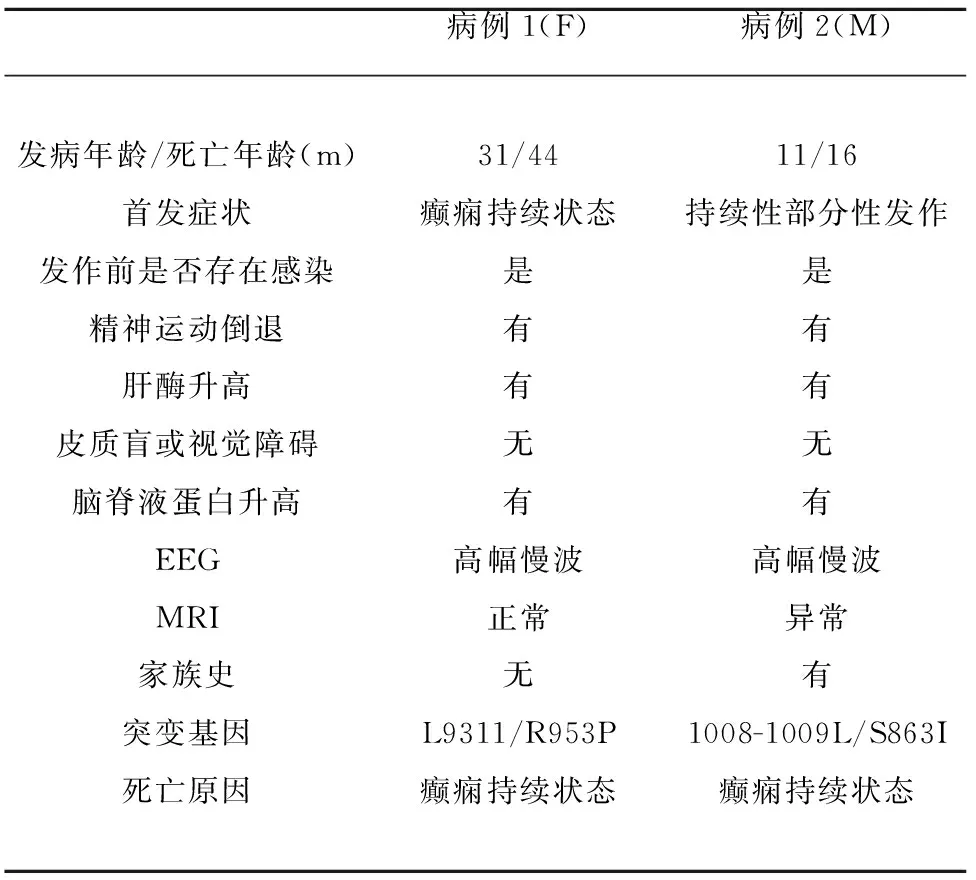
表1 2例病例一般情况
2 讨 论
Alpers综合征(Alpers-Huttenlocher综合征、儿童肝脑退行性病变、弥漫性进行性脑灰质变性综合征)是人们最早认识的线粒体疾病表型中的一种,发病年龄为1个月~25岁,以婴幼儿常见,并多于5岁前发病。其典型四联征表现包括:难治性癫痫;精神运动发育的倒退;进行性肝衰竭,或在应用丙戊酸治疗后出现急性肝衰竭;皮质盲。其他表现包括偏头痛样症状,共济失调,视力障碍,肢体局部的麻痹或颤动,肌阵挛或肢体抽搐等。有报道认为此类患者临床表现的差异主要取决于突变基因的不同[4]。
Alpers综合征作为一种常染色体隐性遗传的肝脑综合征,发病率低,目前尚无具体统计数据,本文对相关文献报道进行总结,归纳了60例资料较为完整患者基本情况。其中除11例无性别记载外,余49例中男性患者24例,女性患者25例。发病年龄最小者1 m,最大者25岁,平均发病年龄为4.5岁,平均死亡年龄为5.9岁。临床表现为视觉异常者24例,占40%。肝酶异常或肝衰竭者52例,占87%,其中3例有使用丙戊酸的记载。生长发育迟缓或精神运动倒退者42例,占70%。癫痫发作者56例,占93%。其中25例进行突变基因检测,其中A467T突变18例,W748S突变6例,L83P突变1例[1,3~11]。
本病以脑皮质受累为主,常伴有枕叶病变,表现为神经元丢失,海绵样变性,胶质细胞增生等,枕叶皮质的普遍受累提示了脑电图、影像学及病理检查方面的相似性。Matthew等指出Alpers综合征患者脑电图基本电节律表现为后头部主导的异常δ慢波节律[5],其认为这些放电类型的不同可能反映了大脑皮质功能倒退的不同时期,而以后头部为主导的慢波节律可能说明代谢活动高的枕部区域可能更易受到疾病的影响。本文报道的2例患者脑电图表现符合类似规律,其中伴有单侧肢体抽动者脑电图表现为对侧皮质癫痫波的发放。该类患者头部CT常表现为双侧枕叶及颞叶低密度影,脑组织的海绵样变性,即表现为弥散性神经元变性或损失和星型细胞增生,以大脑灰质最为严重,白质轻微受累,而基底节、脑桥、脑干和小脑并不受累及[12],也报道本病影像学可存在白质病变[13~15]。
肝衰竭作为此病临床特点之一,却很少能作为本病的诊断标准性测试。因为肝衰竭往往见于疾病后期,临床使用丙戊酸控制癫痫发作的患者发病可能相对较早。肝组织病理检查可有结节增生,肝细胞丢失,胆管增殖,脂肪变,胆汁淤积。本文报道的2例患儿均伴有肝酶升高,但无明确肝实质损伤,保肝治疗可有好转但随病情反复呈反复趋势。诊断明确避免了使用丙戊酸导致急性肝衰竭。脑脊液蛋白及乳酸盐异常是本病的特点之一[6]。本文2例患儿脑脊液蛋白升高符合上述特点。
本病的诊断最终依赖于基因检测。POLG位于15q25,编码线粒体DNA多聚酶γ,该蛋白在线粒体DNA复制与修复中起重要作用。POLG基因突变可以导致不同表型的线粒体疾病,Alpers综合征是其中的一种。突变可以是纯合突变,也可以表现为杂合突变,但更常见的是杂合突变。Nguyen等指出A467T置换是造成本病最常见的基因突变,该基因占据目前造成本病突变基因的40%[13,16,17],其他常见的突变位点有G2899T、G1681T、G2824T、G2525C。
本文报道的2例患儿起病年龄分别为31 m、11 m,1例有相关家族史,均以癫痫持续状态起病,此后癫痫反复多次发作,呈难治性,病初伴有呼吸道感染,1例存在生长发育落后,1例发病后出现运动发育倒退,无皮质盲等眼部病变,实验室检查除感染指标异常,心肌酶、转氨酶轻微升高外,脑脊液蛋白含量增加,脑电图表现为枕部为主的慢波节律,头部MRI或CT无典型临床表现,经基因检测证实为Alpers综合征。诊断明确避免了使用丙戊酸控制癫痫发作引起急性肝衰竭,但2例患儿最终死于癫痫持续状态。本文报道的2例患者突变基因类型目前尚未见临床致病性报道,但此2例患儿的发病年龄、临床表现、相关实验室检查、转归均符合本病特点。需要注意的是,本病虽然是线粒体基因突变导致的疾病,但2例患儿发病前均存在感染因素,临床上亦有此类疾病首次发作与感染相关的报道[6],考虑与感染导致能量代谢旺盛有关。
本病目前尚无有效的治疗手段,多于发病后3 m到12 y内死亡,婴儿期发病的患者很少能够度过青年时期。死亡原因主要是癫痫持续状态和急性肝衰竭。因此对于临床儿科工作者,尤其是小儿神经科医生,应提高对于本病的认识。对于婴幼儿起病,突然出现的难治性癫痫、视觉障碍、肝衰竭或是持续肝酶升高、精神运动发育倒退都应想到本病的可能性,应尽早行基因检查明确。治疗上应避免使用丙戊酸控制癫痫发作,以免引起急性肝功能衰竭。
[1]Harding BN. Progressive neuronal degeneration of childhood with liver disease (Alpers-Huttrnlocher syndrome):A personal review[J]. J Child Neural,1990,5(4):273-287.
[2]Alpers BJ. Diffuse progressive degeneration of the gray matter of the cerebrum[J]. Arch Neurol Psychiatry,1931,25:469-505.
[3]包新华,吴 晔,熊 晖,等. Alpers综合征的临床与病理特点及基因突变分析[J]. 实用儿科临床杂志,2008,23(24):1911-1913.
[4]Uusimaa J,Hinttala R,Rantala H,et al. Homozygous W748S mutation in the POLGI gene in patients with juvenile-onset Alpers syndrome and status epilepticus [J]. Epilepsia,2008,49(6):1038-1045.
[5]Matthew F,Peters H,Salemi R,et al. Alpers syndrome with mutations in POLG:Clinical and investigative features [J]. Pediatric Neurology,2011,45:311-318.
[6]Nguyen KV,Qstergaard E,Holst Ravn S,et al. POLG mutations in Alpers syndrome [J]. Neurology,2005,65(9):1493-1495.
[7]Naviaux RK,Nguyen KV. POLG Mutations associated with Alpers’ Syndrome and mitochondrial DNA depletion [J]. Ann Neurol,2004,55:706-712.
[8]Montine TJ,Powers JM,Vogel FS,et al. Alpers’ syndrome presenting with seizures and multiple stroke-like episodes in a 17-year-old male [J]. Clin Neuropathol,1995,14:322-326.
[9]Harding BN,Alsanjari N,Smith SJ,et al. Progressive neuronal degeneration of childhood with liver disease (Alpers’ disease) presenting in young adults [J]. Neurol Neurosurg Psychiatry,1995,58:320-325.
[10]Worle H,Kohler B,Schlote W,et al. Progressive cerebral degeneration of Childhood with liver diseases (Alpers Huttenlocher disease) with cytochrome oxidase deficiency presenting with epilepsia partialis continua as the first clinical manifestation [J]. Clin Neuropathol,1998,17:63-68.
[11]Klein H,Dichgans J. Familiare juvenile glio-neurale Dystrophie. Akut beginnende progressive Encephalopathie mit rechtsseitigen occipito-parietalen Herdsymptomen and Status epilepticus [J]. Arch Psychiatr Nervenkr,1969,212:400-422.
[12]Guo YP,Guo Z,Liu HW,et al. Progressive neuronal degeneration of childhood with liver’s disease (Alpers’ Disease) clinical features and neuropathological studies of 4 sibling [J]. Chinese Journal of Clinical Neurosciences,2000,8(s1):95.
[13]Tzoulis C,Engelsen BA,Telstad W,et al. The spectrum of clinical disease caused by the A467T and W748S POLO mutations;A study of 26 cases [J]. Brain,2006,129 (7):1685-1692.
[14]Invernizzi F,Varanese S,Thomas A,et al. Two novel POLG1 mutations in a patient with progressive external ophthalmoplegia,levodopa-responsive pseudo-orthostatic tremor and parkinsonism [J]. Neuromuscul Disord,2008,18(6):460-464.
[15]Stuart GR,Santos JH,Strand MK,et al. Mitochondrial and nuclear DNA defects in Saccharomyces cerevisiae with mutations in DNA polymerase gamma associated with progressive external ophthalmoplegia [J]. Hum Mol Genet,2006,15 (2):363-374.
[16]Nguyen KV,Sharief FS,Chan SL,et al. Molecular diagnosis of Alpers syndrome [J]. J Hepatology,2006,45(1):108-116.
[17]Horvath R,Hudson G,Ferrari G,et al. Phenotypic spectrum associated with mutations of the mitochondrial polymerase gamma gene [J]. Brain,2006,129(7):1674-1684.
1003-2754(2017)09-0840-03
R742
2017-5-20;
2017-09-02
(吉林大学白求恩第一医院小儿神经科,吉林 长春 130021)
陈银波,E-mail:ybcyb2010@aliyun. com
猜你喜欢
中国民间疗法(2021年10期)2021-07-22 02:33:12
现代电生理学杂志(2016年1期)2016-07-10 10:20:58
中国卫生标准管理(2015年24期)2016-01-14 09:29:00
中国卫生标准管理(2015年5期)2016-01-14 05:16:56
川北医学院学报(2015年5期)2015-12-05 08:22:33
中国学术期刊文摘(2015年8期)2015-10-29 09:51:18
现代电生理学杂志(2015年1期)2015-07-18 11:02:17
中国当代医药(2015年7期)2015-03-01 02:01:13
现代检验医学杂志(2015年1期)2015-02-06 01:59:29
四川生理科学杂志(2014年2期)2014-02-28 14:09:02
