
白菜类作物开花时间的全基因组关联分析
2017-09-26 08:03高宝祯刘博李石开梁建丽程锋王晓武武剑
中国农业科学 2017年17期
高宝祯,刘博,李石开,梁建丽,程锋,王晓武,武剑
白菜类作物开花时间的全基因组关联分析
高宝祯1,刘博1,李石开2,梁建丽1,程锋1,王晓武1,武剑1
(1中国农业科学院蔬菜花卉研究所,北京 100081;2云南省农业科学院园艺作物研究所,昆明 650205)
【目的】解析白菜类作物开花时间的调控位点,定位白菜类作物开花时间相关的候选基因,为白菜类作物抽薹开花时间的遗传改良提供依据。【方法】以116份白菜类作物组成的自然群体作为研究材料,分别种植在温室与露地2个独立的环境中,进行开花时间调查。同时,提取试验材料的DNA样品进行深度为1.2x的重测序,对测序数据用Pooled Mapping法进行过滤、与参考基因组比对,获得全基因组高密度SNP集合。经过条件过滤后,对高质量的SNP集合进行生物信息分析,包括试验材料的群体结构分析和全基因组连锁不平衡分析。从高质量的SNP集合中,随机挑选出2 000个变异位点,用PhyML软件以最大似然法对116份试验材料进行系统发育树分析。用全部的高质量SNP集合位点通过软件Haploview进行全基因组连锁不平衡分析。最后,将高质量的SNP集合与开花时间数据结合,通过TASSEL和GAPIT软件包以及R程序语言进行全基因组关联分析。根据强关联峰值信号点位置和连锁不平衡区间定位开花时间候选位点,再通过白菜与同源物种拟南芥的基因共线性关系以及基因功能注释分析来预测白菜类作物开花时间相关的候选基因。【结果】不同种植条件下、不同类型的白菜类作物在开花时间上存在广泛差异。试验材料在露地环境下的开花时间高峰期明显早于温室环境下的材料;试验材料在露地环境下的开花时间总体表现出偏正态分布,而在温室环境下,开花时间各个阶段呈现出较为均衡的分布。温室与露地环境下的开花时间呈显著正相关。通过生物信息学分析最终得到的高质量SNP位点共103万个。试验材料的群体结构分析表明在系统发育树上各亚群内部分布较为集中,不同亚群之间的分布与材料的地理起源密切相关。全基因组衰减平均LD为2.3 kb,表明在116份白菜类作物构建的群体内存在较为频繁的重组和突变。对不同条件下的开花时间进行全基因组关联分析,用复合模型检测到54个(>4)强关联峰值信号点,一般模型检测到87个(>5)。通过进一步分析强关联信号点的连锁不平衡(linkage disequilibrium,LD)区段,得到存在强连锁关系(2>0.33)的峰值信号点共33个(温室环境下27个,露地环境下19个)。其中,在温室与露地环境下的共定位位点13个。根据33个关联候选位点,再通过白菜与同源物种拟南芥的基因共线性关系以及基因功能注释分析筛选出白菜类作物开花时间相关的候选基因14个,其中温室与露地环境下共定位候选基因3个(、和)。在露地条件下定位到开花关键基因。【结论】不同条件下开花时间的相关性分析表明,遗传效应在开花早晚中起着决定性作用。全基因组关联分析共鉴定出33个与开花时间相关的显著关联信号。通过连锁不平衡分析、白菜与同源物种拟南芥的基因共线性关系以及基因功能注释分析初步鉴定出14个白菜类作物开花时间相关的候选基因。
白菜类作物;开花时间;连锁不平衡;全基因组关联分析;候选基因
0 引言
【研究意义】白菜类作物()属于十字花科芸薹属(genusof the familyBrassicaceae),是中国乃至世界范围内重要的蔬菜和油料作物,主要包括大白菜、小白菜、芜菁、菜薹、紫菜薹、白菜型油菜等丰富的类型。抽薹开花是白菜类作物生产中很重要的农艺性状。在中国春季及高寒地区的秋冬白菜类作物生产中,先期抽薹开花常常成为降低蔬菜产量和品质的重要难题。因此,挖掘白菜类作物抽薹开花调控位点和基因,培育晚抽薹开花品种,对于保证蔬菜质量和产量、降低菜农损失具有重要意义。【前人研究进展】植物开花是基因网络与环境因素相互作用的结果。植物开花的调控网络在模式植物拟南芥中已有了深入的研究[1-6],目前,已发现有180多个基因与调控抽薹开花有关,且花期调控存在多条调控路径(光周期路径、春化路径、自主路径、赤霉素路径、年龄路径等)。WANG等[7-8]研究认为,虽然白菜类作物与拟南芥具有很近的亲缘关系,但白菜类作物在与拟南芥分化后经历了基因组三倍化事件,复制的开花基因在白菜类作物基因组二倍化过程中有些保留了多拷贝,有些则仍然保持单拷贝,有的则丢失。因而白菜类作物的开花调控相比拟南芥而言更为复杂。前人开展了许多白菜类作物开花时间的分子遗传分析,但主要的研究策略是利用分离群体进行QTL分析,以鉴定出可能的候选基因。LOU等[8]用多种白菜类作物(包括159份高代自交系群体、50份自然群体和8份野生群体材料)进行QTL分析,发现开花时间QTL部分与生物钟重叠;WU等[9]通过对159份白菜类作物序列分析,发现的一个InDel变异与开花时间关联,并在分离群体中验证;XIAO等[10]利用90份DH材料通过转录共表达分析发现是调控白菜类作物开花时间的关键因子;KITAMOTO 等[11]利用白菜类作物F2群体材料开发了SSR标记,发现在第一个内含子的长插入片段可导致晚抽薹。目前,GWAS已经在模式植物拟南芥[12-13]、玉米[14]、水稻[15-16]、大豆[17]等重要农作物中有所应用,并找到了比较可靠的关联信号。随着各物种全基因组测序的完成,大规模重测序的开展和SNP标记的大量开发,利用GWAS研究植物复杂数量性状成为强有力的工具。【本研究切入点】QTL定位的方法仅能对在分离群体的亲本材料间存在差异的基因的效应进行分析,无法在全基因组范围广泛挖掘参与开花调控的基因。而白菜类作物开花时间的GWAS研究尚未见报道。【拟解决的关键问题】采用GWAS鉴定目标性状显著关联SNP,根据连锁不平衡强度和基因注释信息,确定控制性状变异的候选基因。为分子标记辅助选择育种,培育晚抽薹开花品种提供理论依据。
1 材料与方法
室内试验和分析于2015—2016年在中国农业科学院蔬菜花卉研究所完成。
1.1 试验材料
供试材料共116份,来自于国内外广泛收集的代表性白菜类作物资源,大白菜、小白菜、菜薹、紫菜薹、白菜型油菜主要来自中国和日本,芜菁、意大利菜心主要来自欧洲,黄籽沙逊油菜来自南亚(表1)。所有材料均为自然群体,所有材料的名称、编号、属性及来源见电子附表1。
1.2 种植处理
试验1:2009年10月至2010年4月,将116份试验材料种植于云南省农业科学院试验基地的露地大田。试验2:2010年10月至2011年3月,将116份试验材料种植于中国农业科学院蔬菜花卉研究所的玻璃温室。2个独立试验均在25℃催芽,出芽后播种于穴盘育苗。播种30 d后,分别定植到露地(试验1)和塑料花盆(10 cm×10 cm)(试验2)中。试验2中花盆内的土壤为3﹕1的草炭﹕蛭石。栽培过程中采用常规管理,视需要适当施肥,无其他特殊处理。每份材料设置4个重复单株。
1.3 开花时间调查
从播种到出现第一朵花所需的时间作为开花时间(days to flowering,DTF)。每日对开花情况进行记录。待调查试验结束后,取每份材料的花期平均值作为表型数据,进行关联分析。根据调查截止时间,将未开花的材料的开花时间定为150 d。
1.4 全基因组重测序
选取苗期的白菜幼嫩叶片,采用改良CTAB法提取每一份材料的DNA样品。取样时对温室环境下的每份材料选1个单株取样。提取的DNA样品采用Illumina HiSeqTM2000 平台(Illumina Inc., San Diego, CA, USA)进行双末端重测序,产生100 bp长的pair-end(PE)reads。116份重测序材料的总数据量为77G,平均每个样本的测序深度为1.2x。
1.5 重测序分析与基因分型
将116份材料的reads集合利用FU等[18]的方法对低深度重测序进行过滤、参考基因组比对,得到单核苷酸多态性变异(SNP)集合。采用临近算法(KNN)[16]对SNP集合的缺失数据进行填补处理。
对填补后的数据进行过滤:(1)过滤掉基因型缺失度超过70%的变异位点;(2)过滤掉最小等位基因频率(MAF)<0.05的变异位点;(3)只保留二态的变异位点。过滤后得到高质量的SNP矩阵用于后续分析。
1.6 群体结构分析
从上述SNP集合中,利用随机函数挑选出2 000个变异位点进行如下分析:采用PhyML version 3.0[19]软件以默认参数对116份白菜类作物进行系统发育分析(最大似然法,maximum likelihood)。
1.7 全基因组连锁不平衡分析
用过滤后SNP集合位点进行全基因组连锁不平衡(linkage disequilibrium,LD)分析[20],分析采用软件Haploview[21]进行。
1.8 全基因组关联分析
将花期调查的表型数据和过滤后构建的高质量SNP矩阵,通过一般线性模型(general linear model,GLM)和压缩复合线性模型(compressed mixed linear model,cMLM)分别对116份白菜类作物的花期性状进行全基因组关联分析。GLM使用软件TASSEL version 3.0[22],cMLM使用GAPITR软件包[23]。
1.9 候选基因预测
根据GWAS结果获得的强关联信号(一般线性模型>5;压缩复合线性模型>4)和白菜基因组LD区段,来确定强选择位点处LD区段上的白菜开花时间的候选基因,再结合白菜的近缘物种拟南芥与白菜基因的共线性分析结果和功能注释信息(database,http://brassicadb.org /brad/),从而找到开花相关的候选基因。
2 结果
2.1 白菜类作物开花时间表型数据
在温室和露地大田对116份试验材料进行开花时间调查,结果表明,白菜类作物在开花时间上存在广泛的差异(表1),温室条件下开花时间的变异范围为46—150 d;大田条件下开花时间的变异范围为52—150 d。在不同类型的白菜类作物间开花时间也存在差异,芜菁开花普遍最晚,结球白菜次之,非结球白菜类(小白菜、意大利菜心、菜薹、紫菜薹、乌塌菜)较早,而白菜型油菜的开花时间则早晚均有,这与其包含春油菜和冬油菜有关。
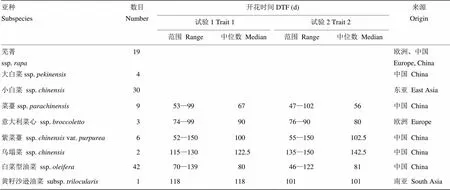
表1 试验材料类型及其在露地和温室的开花时间

图1 116份白菜类作物开花时间的频数分布图
由图1可知,在41—60 d中,露地环境下只有3份材料开花,温室环境下有17份;61—80 d和81—100 d中,露地环境下的材料开花数目达到高峰,分别有26、28份材料开花,而温室环境下则分别有18、20份材料开花;在101—120 d和121—140 d内,温室环境与露地环境的材料开花数目差异不明显。这说明试验材料在不同环境中开花时间存在差别。在露地环境下61—80 d和81—100 d中开花的材料,在温室环境下的开花时间提早至41—60 d;露地环境下的材料开花高峰期却明显早于温室环境下的材料;试验材料在露地环境下的开花时间总体表现出偏正态分布,而在温室环境下,开花时间各个阶段呈现出较为均衡的分布。
对温室与露地环境下的开花时间做相关性分析(图2),结果表明,2个环境下的开花时间之间存在高度正相关关系,相关系数为0.89,说明遗传效应在开花早晚中起着决定性作用。
2.2 群体结构分析及全基因组连锁不平衡分析
由于样本的全基因组重测序覆盖度较低,平均为1.2x左右,因此,导致一些样本的基因型缺失,通过KNN算法,对缺失基因型进行了填补。经过填补和过滤后,最终得到高质量的SNP集合共有103万个SNP变异位点。
从上述SNP集合中随机挑选出2 000个SNP位点进行了系统发育树分析(图3),结果显示,同为欧洲-地中海一带起源的芜菁和意大利菜心在发育树上更为接近,而主要起源于东亚、同属非结球白菜类的小白菜、菜薹、紫菜薹、乌塌菜在发育树上更为接近,白菜型油菜则分到2个明显的分支上。表明116份白菜类试验材料在系统发育树上各亚群内部分布较为集中,不同亚群之间的分布与材料的地理起源密切相关。

图2 温室与露地环境下开花时间的相关性

粉红:非结球白菜类(小白菜、菜薹、紫菜薹、乌塌菜);绿色:白菜型油菜(春油菜、冬油菜、黄籽沙逊油菜);红色:大白菜;蓝色:欧洲类型(芜菁、意大利菜心) Pink: Non-heading leafy type (ssp. chinensis, ssp. parachinensis, ssp. chinensis var. purpurea, ssp. chinensis); Green: Oilseed type (spring oilseed, winter oilseed, subsp. trilocularis); Red: ssp. pekinensis; Dark blue: ssp. rapa; Light blue: ssp. broccoletto
用上述全部SNP位点进行全基因组连锁不平衡分析(图4),可知,当2从0.48衰减到0.26时,所对应的物理距离为2.3 kb,因此,白菜类作物的全基因组平均LD为2.3 kb(当2值衰减到其最大值一半时对应的物理长度,即为全基因组平均LD)。
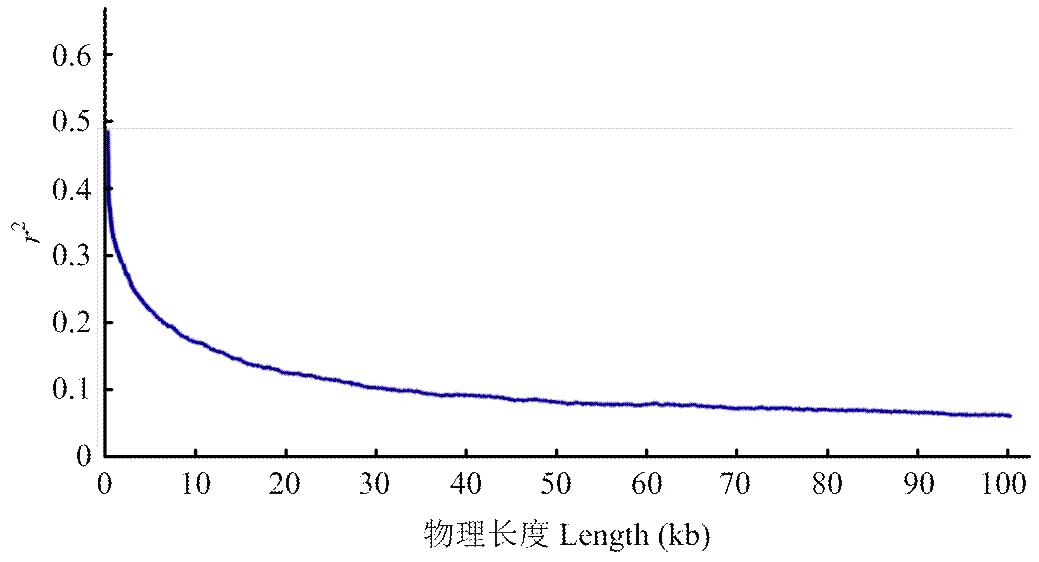
图4 116份白菜类作物全基因组连锁不平衡衰减
分别利用GLM和cMLM模型对开花时间进行全基因组关联分析(图5)。对获得的显著信号点进行连锁强度分析过滤,得到存在强连锁关系(2>0.33)的峰值信号点共33个(温室环境下27个,露地环境下19个)。其中,在温室与露地环境下的共定位位点有13个(表2)。
对33个强关联信号位点的功能注释信息分析(表2)可知,有21个位点位于基因间区,4个点位于内含子,7个点位于外显子区域,其中只有2个点(SNP=A02_25176329,SNP=A09_26670099)是非同义突变,前者由异亮氨酸(Ile)变为苏氨酸(Thr),后者由丙氨酸(Ala)变为丝氨酸(Ser),两者都发生了氨基酸极性性质的改变,这可能会引起其编码蛋白发生较大的功能变异。
2.3 候选基因预测
通过温室与露地环境下定位到的开花时间强关联信号位点,分析其所在的连锁不平衡区段,同时,根据白菜与拟南芥基因的共线性分析与功能注释结果(database,http://brassicadb.org/brad/),共定位到14个白菜花期控制的候选基因与拟南芥存在共线性同源关系,同时,对候选基因进行了GO注释分析(电子版附表2)。14个白菜开花时间候选基因中,温室与露地环境下共定位3个候选基因(、和),在露地条件下定位到开花时间关键因子(表3)。

A:温室试验_一般模型;B:温室试验_复合模型;C:露地试验_一般模型;D:露地试验_复合模型
3 讨论
随着测序技术的发展,基因组测序和重测序已经在重要农作物研究中普及应用,通过全基因组关联分析(GWAS)来挖掘目标性状的候选基因成为可能。GWAS比传统QTL研究有明显的优势,且已在多个模式植物和重要农作物中得到应用[12,15,17]。
开花时间是影响白菜类作物产量和质量的重要农艺学性状,因此,利用GWAS分析挖掘白菜类作物晚开花基因对于白菜耐先期抽薹育种具有重要的价值。目前关于白菜类作物开花时间变异的关联分析还未见报道。白菜类具有非常丰富的亚群,各亚群的开花时间存在很大差异。本研究选取了来自于东亚、欧洲、南亚的白菜类作物材料,尽可能覆盖了白菜类作物栽培的不同地域,构建了一个具有丰富开花时间变异的自然群体材料作为试验样本,保证了表型数据的多样性。此外,本研究通过全基因组低乘数重测序的方法,获得了大量的SNP标记,数量级达到百万,且经过严格的过滤筛选,在达到全基因组分布密度的基础上保证了标记的可靠性。并采用了2个环境对关联结果进行LD连锁强度(2>0.33)筛选,进一步提高了关联分析的准确性。
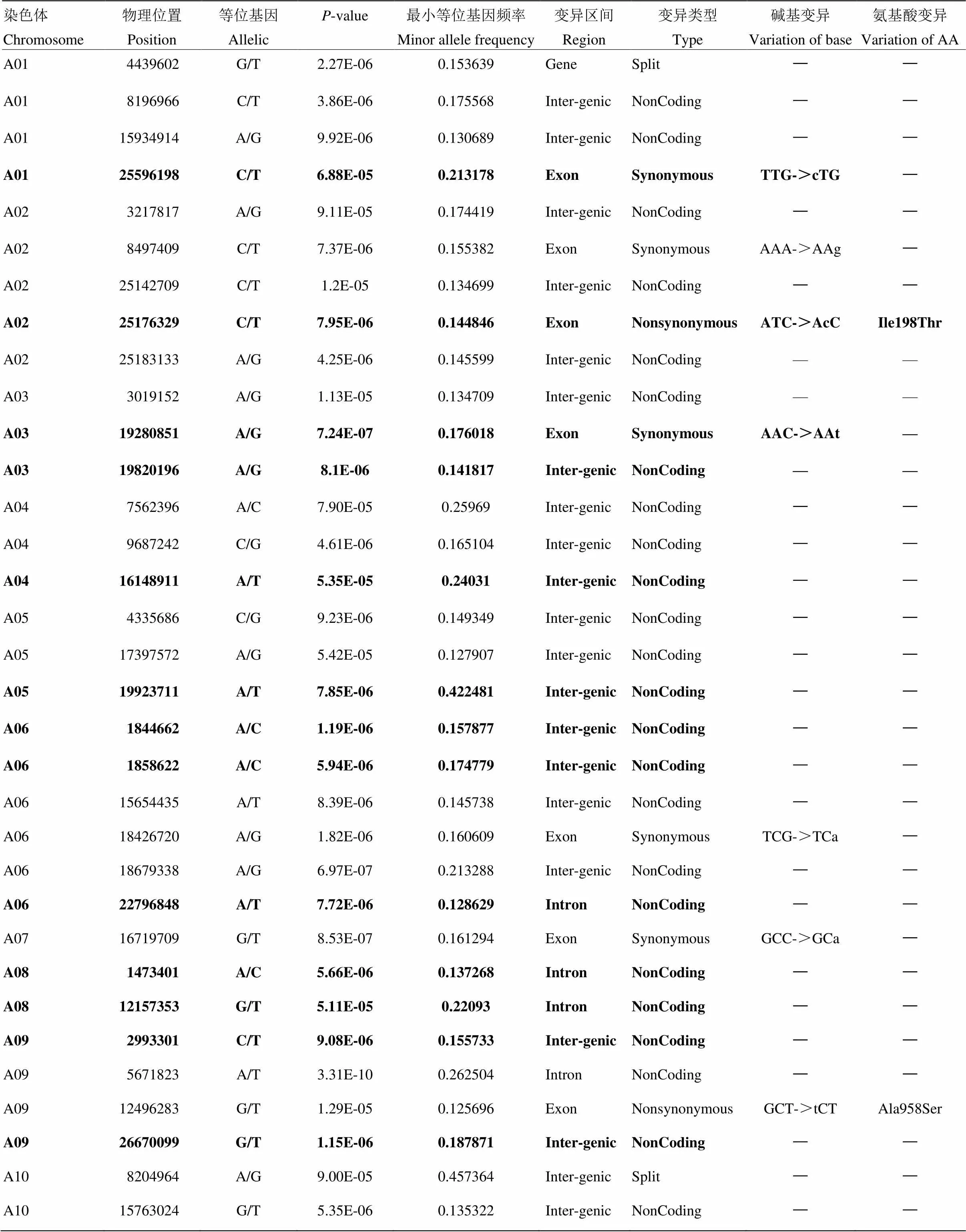
表2 强关联信号位点及注释信息
粗体表示温室与露地环境下的13个共定位位点信息
Bold font refers to information of 13 co-identity signals under greenhouse and open-field environment

表3 白菜开花时间的候选基因
BARCHI等[24]报道拟南芥184份材料组成的自然群体在露地和严格控制环境条件的温室中开花时间的相关性低,并由此得出在自然条件而非严格控制的环境条件下研究开花时间的基因型与环境互作非常重要。本研究的表型调查分别在日光温室和露地条件下进行,均为白菜生产上的栽培条件,以期获得可以用于耐抽薹育种的位点或分子标记。研究结果显示,116份材料在2种不同的栽培条件下的开花时间显著相关,并且通过GWAS分析共鉴定出33个强关联信号点,其中13个是在露地和温室条件下共定位的关联信号。
本研究中有34份材料属于高代自交系材料(电子版附表1),付丽霞[25]对包含这批自交系材料的白菜类作物进行了低深度重测序材料的CAPS/dCAPS分型验证。结果显示,通过本研究中的Pooled Mapping法获得的位点基因型与CAPS/dCAPS试验的分型结果完全一致。验证的位点一致性说明Pooled Mapping法在低深度重测序中进行SNPs分型结果具有可靠性。同样的基因分型方法应用于甘蓝的低深度测序中,验证结果同样证明了Pooled Mapping法具有可靠性[18]。
利用白菜与拟南芥的共线性关系[26],本研究在强信号关联位点共找到14个白菜开花时间相关的候选基因,其中,既包括已报道的开花调控基因如开花信号整合因子(),也包括一些功能未知的基因。这些基因对于指导白菜耐抽薹性选育具有参考价值。
本研究中,温室与露地环境下共定位到3个候选基因(、和)。在模式植物拟南芥中,已经被证实具有促进开花的作用,在开花通路中参与到开花应答与组织发育路径中[27-29]。MELZER等[27]研究发现,不仅能调控开花时间,而且影响组织确定性。FUL蛋白活性可以有效增加植物寿命而不受开花时间影响。Teper-Bamnolker等[28]发现,开花时间调控的开关基因可以调控在叶片中的积累。在组织中的积累与开花转变相关,并且对其具有促进作用。Ferrándiz等[29]研究发现,当作用元件MADS-box变异与和一起出现时,会导致其突变体植株不开花,而在开花位置不断地生成叶片分生组织。此外,研究发现,小麦中的与拟南芥中的有高度同源性,且在分生组织发育过程中与花期调控相关[30]。虽然目前在白菜类作物开花调控中还未见相关报道,但是根据Melzer等[27]的研究,不管环境条件如何影响开花时间,都能够参与到组织发育过程中,由此推测,在本研究中,虽然温室和露地环境的差异造成了开花时间上的差异,但是却都定位到了。因此,结合目前已知作物的研究可推测其在白菜类作物中参与到花期调控的组织发育阶段和路径中。
在拟南芥的开花调控中参与到光周期路径中,与红光/远红光的光受体作用有关。光周期路径中,植物开花不仅受光照长度影响,还受到光谱影响。PHYB蛋白可以作为光受体吸收红光/远红光,促进CO蛋白降解,抑制表达,从而延迟开花,其突变体在长/短日照下都能表现出早开花[31-33]。低强度或低频率的红光/远红光会降低PHYB蛋白光受体(光敏色素B)的活性,这使得PHYTOCHROME INTERACTING FACTORS(PIFs)直接激活生长素合成基因的翻译表达,而引起避光反应,包括拟南芥早开花和茎部伸长[34]。前人对植物中光受体信号网络的研究发现,不同的物种中,PHYB蛋白拥有其他伙伴成员,但是PHYB是最重要的一类与遮阴反应有关的光受体。拟南芥中有PHYB、PHYD、PHYE[35-38],番茄中有PHYB、PHYB2、PHYE[39],还有黄瓜[40]、白菜[41-42]、高粱[43]等都含有PHYB蛋白受体[44]。Robson等[45]对拟南芥和白菜作物的突变进行了研究,的变异可降低感受红光﹕远红光的比例。由此推测,白菜中的候选基因可能参与到光周期路径中。
目前,关于的研究主要集中于拟南芥中,有报道指出,通过赤霉素(GA)信号途径,拟南芥在长/短日照下都能引起早开花[46-47]。可以改变水稻的开花时间和根部发育状况[48]。黄琼华等[49]将拟南芥早花基因通过农杆菌介导法转化获得了再生转基因植株,受体为白菜类作物的近源物种甘蓝型油菜的下胚轴。但是没有进一步研究其对转基因甘蓝型油菜开花时间的影响。
另外,本研究中,白菜类作物的连锁不平衡衰减距离为2.3 kb,与最新的研究结果[50](~3 kb)相近,小于拟南芥基因组的LD衰减率[51](~10 kb),而远远小于水稻基因组平均LD[16](>100 kb)。这可能与白菜类作物是异花授粉植物,具有自交不亲和性有关,说明在116份白菜类作物构建的群体内存在较为频繁的重组和突变。
GWAS可以作为白菜类作物开花时间候选基因定位的有力工具,通过这一分析获得的候选基因为分子辅助育种提供依据,下一步将深入开展上述候选基因的功能研究,进一步揭示开花时间调控的分子遗传机理,为选育白菜类作物耐抽薹品种打下基础。
4 结论
GWAS分析检测到33个与白菜类作物开花时间显著关联的SNP信号(温室环境下27个,露地环境下19个),其中温室与露地环境下的共定位位点13个。白菜与同源物种拟南芥的基因共线性关系以及基因功能注释分析初步鉴定出14个白菜类作物开花时间相关的候选基因。
References
[1] ROUX F, TOUZET P, CUGUEN J, LE C V. How to be early flowering: an evolutionary perspective.,2006, 11(8): 375-381.
[2] ALONSO-BLANCO C, KOORMNEEF M. What has natural variation taught us about plant development, physiology, and adaptation?,2009, 21(7): 1877-1896.
[3] KIM D H, DOYLE M R, SUNG S, AMASINO R M. Vernalization: winter and the timing of flowering in plants.,2009, 25(1): 277-299.
[4] FORNARA F, DE M A, COUPLAND G. Snaphot: Control of flowering in.,2010, 141(3): 1-2.
[5] WAHL V, PONNU J, SCHLERETH A, ARRIVAULT S, LANGENECKER T, FRANKE A, FEIL R, LUNG JE, STITT M, SCHMID M. Regulation of flowering by trehalose-6-phosphate signaling in.,2013, 339(6120): 704-707.
[6] SHRESTHA R, GOMEZ-ARIZA J, BRAMBILLA V, FORMARA F. Molecular control of seasonal flowering in rice,and temperate cereals.,2014, 114(7): 1445.
[7] WANG X, WANG H, WANG J, SUN R, WU J, LIU S, BAI Y, MUN J H, BANCROFT I, CHENG F,. The genome of the mesopolyploid crop species., 2011, 43(10): 1035-1039.
[8] LOU P, XIE Q, XU X, EDWARDS C E, BROCK M T, WEINIG C, MCCLUNG C R. Genetic architecture of the circadian clock and flowering time in.,2011, 123(3): 397-409.
[9] WU J, WEI K, CHENG F, LI S, WANG Q, ZHAO J, BONNERMA G, WANG X. A naturally occurring InDel variation in BraA.FLC.b (BrFLC2) associated with flowering time variation in.,2012, 12(1): 1-9.
[10] XIAO D, ZHAO J J, HOU X L, BASNET R K, CARPIO D P D, ZHANG N W, JOHAN B, KE L, FENG C, Wang X W. Thehomologueis a key regulator of flowering time, identified through transcriptional co-expression networks.,2013, 64(14): 4503-4516.
[11] KITAMOTO N, YUI S, NISHIKAWA K, TAKAHATA Y, YOKOI S. A naturally occurring long insertion in the first intron in theFLC2 gene causes delayed bolting.,2014, 196(2): 213-223.
[12] ATWELL S, HUANG Y S, VILHJALMSSON B J, WILLEMS G, HORTON M, LI Y, MENG D, PLATT A, TARONE A M, HU T T,. Genome-wide association study of 107 phenotypes ininbred lines.,2010, 465(7298): 627-631.
[13] BRACHI B, MORRIS G P, BOREVITZ J O. Genome-wide association studies in plants: the missing heritability is in the field.,2011, 12(10): 1-8.
[14] WANG M, YAN J, ZHAO J, SONG W, ZHANG X, XIAO Y, ZHENG Y. Genome-wide association study (GWAS) of resistance to head smut in maize., 2012, 196(11): 125-131.
[15] ARANZANA M J, KIM S, ZHAO K, BAKKER E, HORTON M, JAKOB K, LISTER C, MOLITOR J, SHINDO C, TANG C. Genome- wide association mapping inidentifies previously known flowering time and pathogen resistance genes.,2005, 1(5): e60.
[16] HUANG X, WEI X, SANG T, ZHAO Q, FENG Q, ZHAO Y, LI C, ZHU C, LU T, ZHANG Z. Genome-wide association studies of 14 agronomic traits in rice landraces.2010, 42(11): 961-967.
[17] ZHANG J, SONG Q, CREGAN P B, NELSON R L, WANG X, WU J, JIANG G L. Genome-wide association study for flowering time, maturity dates and plant height in early maturing soybean () germplasm.,2015, 16(1): 1-11.
[18] FU L X, CAI C C, CUI Y N, WU J, LIANG J L, CHENG F, WANG X W. Pooled mapping: an efficient method of calling variations for population samples with low-depth resequencing data.,2016, 36(4): 1-12.
[19] GUINDON S, GASCUEL O. PhyML-A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood.,2003, 52(5): 696-704.
[20] YANG J, LEE S H, GODDARD M E, VISSCHER P M. GCTA: A tool for genome-wide complex trait analysis.,2011, 88(1): 76-82.
[21] BARRETT J C, FRY B, MALLER J, DALY M J. Haploview: Analysis and visualization of LD and haplotype maps.,2005, 21(2): 263-265.
[22] BRADBURY P J, ZHANG Z, KROON D E, CASSTEVENS T M, RAMDOSS Y, BUCKLER E S. TASSEL: Software for association mapping of complex traits in diverse samples.,2007, 23(19): 2633-2635.
[23] ZHANG Z, ERSOZ E, LAI C Q, TODHUNTER R J, TIWARI H K, GORE M A, BRADBURY P J, YU J, ARNETT D K, ORDOVAS J M. Mixed linear model approach adapted for genome-wide association studies.,2010, 42(4): 355-360.
[24] BRACHI B, FAURE N, HORTON M, FLAHAUW E, VAZQUEZ A, NORDBORG M, BERGELSON J, CUGUEN J, ROUX F. Linkage and association mapping offlowering time in nature.,2010, 6(5): e1000940.
[25] 付丽霞. 芸薹属蔬菜低深度测序SNP分型及其应用[D]. 北京: 中国农业科学院, 2016.
FU L X. SNP genotyping using low-depth resequencing data and its application invegetables [D]. Beijing: Chinese Academy of Agricultural Science, 2016. (in Chinese)
[26] CHENG F, FANG L, WANG X W. Syntenic gene analysis betweenand otherspecies., 2012, 3: 198.
[27] MELZER S, LENS F, GENNEN J, VANNESTE S, ROHDE A, BEECKMAN T. Flowering-time genes modulate meristem determinacy and growth form in., 2008, 40(12): 1489-1492.
[28] TEPER-BAMNOLKER P, SAMACH A. The flowering integrator FT regulates SEPALLATA3 and FRUITFULL accumulation inleaves., 2005, 17(10): 2661-2675.
[29] FERRáNDIZ C, GU Q, MARTIENSSEN R, YANOFSKY M F. Redundant regulation of meristem identity and plant architecture by FRUITFULL, APETALA1 and CAULIFLOWER., 2000, 127(4): 725-734.
[30] SHIMADA S, OGAWA T, KITAGAWA S, SUZUKI T, IKARI C, SHITSUKAWA N, MURAI K. A genetic network of flowering-time genes in wheat leaves, in which an APETALA1/FRUITFULL-like gene,, is upstream of FLOWERING LOCUS T., 2009, 58(4): 668-681.
[31] ENDO M, NAKAMURA S, ARAKI T, MOCHIZUKI N, NAGATANI A. Phytochrome B in the mesophyll delays flowering by suppressing FLOWERING LOCUS T expression invascular bundles., 2005, 17(7): 1941-1952.
[32] VALVERDE F, MOURADOV A, SOPPE W, RAVENSCROFT D, SAMACH A, COUPLAND G. Photoreceptor regulation of CONSTANS protein in photoperiodic flowering., 2004, 303(5660): 1003-1006.
[33] CERDAN P D, CHORY J. Regulation of flowering time by light quality., 2003, 423(6942): 881-885.
[34] REED J W, NAGPAL P, POOLE D S, FURUYA M, CHORY J. Mutations in the gene for the red/far-red light receptor phytochrome B alter cell elongation and physiological responses throughoutdevelopment., 1993, 5(2): 147-157.
[35] CASSON S A, FRANKLIN K A, GRAY J E, GRIERSON C S, WHITELAM G C, HETHRINGTON A M. Phytochrome B and PIF4 regulate stomatal development in response to light quantity., 2009, 19: 229-234.
[36] LEIVAR P, MONTE E, Al-SADY B, CARLE C, STORER A, ALONSO J M, ECKER J R, QUAIL P H. Thephytochrome-interacting factor PIF7, together with PIF3 and PIF4, regulates responses to prolonged red light by modulating phyB levels., 2008, 20: 337-352.
[37] RAUSENBERGER J, HUSSONG A, KIRCHER S, KIRCHENBAUER D, TIMMER J, NAGY F, SCHAFER E, FLECK C. An integrative model for phytochrome B mediated photomorphogenesis: From protein dynamics to physiology., 2010, 5: e10721.
[38] SWEERE U, EICHENBERG K, LOHRMANN J, MIRA-RODADO V, BAURLE I, KUDLA J, NAGY F, SCHAFER E, HARTER K. Interaction of the response regulator ARR4 with phytochrome B in modulating red light signaling., 2001, 294: 1108-1111.
[39] CAGNOLA J I, PLOSCHUK E, BENECH-ARNOLD T, FINLAYSON S A, CASAL J J. Stem transcriptome reveals mechanisms to reduce the energetic cost of shade-avoidance responses in tomato., 2012, 160: 1110-1119.
[40] ADAMSE P, JASPERS P, KENDRICK R E, KOORNNEEF M. Photomorphogenetic responses of a long hypocotyl mutant ofL.., 1987, 127(5): 481-491.
[41] DEVLIN P F, ROOD S B, SOMERS D E, QUAIL P H, WHITELAM G C. Photophysiology of the elongated internode (ein) mutant ofein mutant lacks a detectable phytochrome B-like polypeptide., 1992, 100(3): 1442-1447.
[42] PROCKO C, CRENSHAW C M, LJUNG K, NOEL J P, CHORY J. Cotyledon-generated auxin is required for shade-induced hypocotyl growth in., 2014, 165(3): 1285-1301.
[43] FINLAYSON S A, Lee I J, MORGAN P W. Phytochrome B and the regulation of circadian ethylene production in sorghum., 1998, 116: 17-25.
[44] FRANKLIN KA, WHITELAM G C. Phytochromes and shade- avoidance responses in plants., 2005, 96: 169-175.
[45] ROBSON P R H, WHITELAM G C, SMITH H. Selected components of the shade-avoidance syndrome are displayed in a normal manner in mutants ofanddeficient in phytochrome B., 1993, 102(4): 1179-1184.
[46] KANIA T, RUSSENBERGER D, PENG S, APEL K, MELZER S. FPF1 promotes flowering in., 1997, 9(8): 1327-1338.
[47] MELZER S, KAMPMANN G, CHANDLER J. FPF1 modulates the competence to flowering in, 1999, 18(4): 395-405.
[48] XU M L, JIANG J F, GE L, XU Y Y, CHEN H, ZHAO Y, BI Y R, WEN J Q, CHONG K. FPF1 transgene leads to altered flowering time and root development in rice., 2005, 24(2): 79-85.
[49] 黄琼华, 杨光伟. 农杆菌介导法将 FPF1 基因导入油菜的研究初报. 西南农业大学学报, 2002, 24(2): 124-127.
HUANG Q H, YANG G W. Preliminary study onmediated transformation of FPF1 gene intoL.., 2002, 24(2): 124-127. (in Chinese)
[50] CHENG F, SUN R, HOU X, ZHENG H K, ZHANG F L, ZHANG Y Y, LIU B, LIANG J L, ZHUANG M, LIU Y X,. Subgenome parallel selection is associated with morphotype diversification and convergent crop domestication inand.,2016, 48(10): 1218-1224.
[51] KIM S, PLAGNOL V, HU T T, TOOMAJIAN C, CLARK R M, OSSOWSKI S, ECKER J R, WEIGEL D, NORDBORG M. Recombination and linkage disequilibrium in., 2007, 39(9): 1151-1155.
(责任编辑 李莉,岳梅)
附表1 本研究所用的植物材料及其开花时间表型
Table S1 Plant materials used in this study and their flowering time phenotype data

编号Code亚群Sub-population品种名称accessions name类型Type试验1Trait 1试验2 Trait 2 开花时间DTF (d)开花时间 DTF (d) NE001芜菁Turnip CGN07164高代自交系 AIL150±0.00150±0.00 NE002芜菁Turnip CGN11010高代自交系 AILNA150±0.00 NE004芜菁TurnipCGN15201高代自交系 AIL95±1.0099±0.63 NE007芜菁TurnipCGN19961高代自交系 AILNA123±1.26 NE009芜菁TurnipCGN06709高代自交系 AIL150±0.00150±0.00 NE010小白菜Pak ChoiUnknown高代自交系 AIL100±1.20121±2.37 NE013芜菁TurnipCGN20735高代自交系 AIL149±1.40150±0.00 NE014芜菁TurnipHong Man Jing高代自交系 AIL150±0.00150±0.00 NE015芜菁TurnipPu Da Cuo Wu Jing高代自交系 AIL150±0.00149±0.15 NE016芜菁TurnipKu Wu Jing高代自交系 AILNA148±0.50 NE017芜菁TurnipHong Yuan Man Jing高代自交系 AIL150±0.00150±0.00 NE019芜菁TurnipMian Tian Huang Man Jing高代自交系 AILNA150±0.00 NE020芜菁TurnipHuang Chang Man Jing高代自交系 AIL147±2.20149±1.25 NE021芜菁TurnipDan Huang Man Jing高代自交系 AIL143±3.50148±1.46 NE022芜菁TurnipTe Hao Chi Man Jing高代自交系 AIL140±8.70150±0.00 NE023芜菁TurnipZi Yuan Man Jing高代自交系 AIL129±1.26150±0.00 NE024大白菜Chinese cabbageZ16DH134±1.20144±0.97 NE025大白菜Chinese cabbageVO2A0014高代自交系 AIL136±0.50147±0.60 NE026大白菜Chinese cabbageV02A0555DH99±1.5569±1.80 NE027大白菜Chinese cabbageCGN07201高代自交系 AIL124±0.70109±1.15 NE030小白菜Pak ChoiVO2B0613高代自交系 AIL129±1.41138±1.55 NE031小白菜Pak Choi225 Bai Cai-Vs-2DH110±0.0097±0.83 NE032小白菜Pak ChoiVO2B0697高代自交系 AIL103±0.5056±0.06 NE033小白菜Pak ChoiVO2B1223高代自交系 AILNA107±3.01 NE034小白菜Pak ChoiShang Hai Wu Yue ManDH103±1.5574±2.32 NE035小白菜Pak ChoiShang Hai Si Yue ManDH149±0.70148±1.38 NE036小白菜Pak ChoiHua Guan Qing Geng Bai CaiDH150±0.00148±0.89 NE037小白菜Pak ChoiFang Zhen Hei You Yi HaoDH118±1.55129±1.43 NE038小白菜Pak ChoiJin Sha Qing Jiang Bai CaiDH134±1.41132±3.46 NE039小白菜Pak ChoiLv Xin Qing CaiDH136±2.50147±1.55 NE040小白菜Pak ChoiSi Ji Xiang Cui Xiao Bai CaiDH111±2.0688±2.06 NE041小白菜Pak ChoiRi Ben Shu Yao Da Tou Qing Jiang BaiDH120±5.07137±4.00 NE042小白菜Pak Choi1263C IIDH120±0.82120±2.60 NE043小白菜Pak ChoiTai Wan Da Tou Qing Jiang Bai CaiDH140±5.35118±2.50 NE044小白菜Pak ChoiXiu Li Piao Er BaiDH129±1.16132±2.06 NE045小白菜Pak ChoiV02B0002DH111±1.74121±1.60 NE046芜菁TurnipUnknownDH124±4.00127±1.73 NE047小白菜Pak ChoiHei You Bai CaiDH131±1.41132±0.05 NE048小白菜Pak ChoiV02B0591DH136±4.92150±0.00 续附表1 Continued table S1 编号Code亚群Sub-population品种名称accessions name类型Type试验1Trait 1试验2 Trait 2 开花时间DTF (d)开花时间 DTF (d) NE049小白菜Pak ChoiV02B0544DH149±0.20135±0.60 NE051小白菜Pak ChoiJing Guan Wang Qing Geng Cai ZhongDH127±0.07122±0.70 NE052小白菜Pak ChoiShang Hai QingDHNA130±1.05 NE053小白菜Pak ChoiGao Jiao Si Ji Qing Bai CaiDH104±1.56101±1.45 NE054小白菜Pak ChoiHuang Yang Xiao Bai CaiDH117±2.30128±2.66 NE055小白菜Pak ChoiHuang Xin Wu高代自交系 AIL122±1.15126±1.55 NE056小白菜Pak ChoiHuang Xin Wu高代自交系 AIL110±2.5292±2.03 NE057小白菜Pak ChoiHuang Xin Wu Bai Cai高代自交系 AIL122±1.50128±1.50 NE058菜心CaixinV02D0130DH83±2.06102±1.58 NE059菜心CaixinV02D0136DH67±4.4047±4.00 NE060菜心CaixinV02D0172DH99±1.1799±0.99 NE061菜心CaixinV02D0212DH62±2.3256±2.30 NE063菜心CaixinV02D0152DH84±0.8767±1.00 NE064芜菁TurnipUnknownDH122±2.00119±1.77 NE066菜心CaixinLiu Ye 70 Tian You Qing Cai XinDH53±2.8953±3.00 NE067菜心CaixinChuan Shan Liu Ye Cai XinDHNA63±0.77 NE068菜心CaixinSi Jiu Cai Xin L58高代自交系 AILNA53±1.69 NE069意大利菜心BroccolettoCGN06825DH99±1.7376±1.50 NE070菜心CaixinUnknownDH52±3.7050±4.00 NE071意大利菜心BroccolettoCGN17278高代自交系 AIL74±3.5690±3.52 NE072意大利菜心BroccolettoCGN15190高代自交系 AIL90±1.2980±1.56 NE073紫菜薹Zi CaitaiW55DH103±3.58111±3.47 NE074紫菜薹Zi CaitaiW184高代自交系 AIL149±0.50150±0.00 NE076紫菜薹Zi CaitaiV02D0107DH97±0.0093±0.06 NE077紫菜薹Zi CaitaiV02D0140DH108±0.71126±0.55 NE078紫菜薹Zi CaitaiV02D0194DH52±1.3255±1.43 NE079紫菜薹Zi CaitaiGai Liang Shi Yue HongDH92±2.6194±2.03 NE083乌塌菜WutacaiLv Ling Wu Ta CaiDH115±3.28135±2.98 NE084乌塌菜WutacaiZhong Ba Ye Wu Ta CaiDH130±0.28150±0.00 NE087油用白菜 OilseedCGN06834高代自交系 AIL139±1.50121±1.37 NE088油用白菜 OilseedCGN06837高代自交系 AIL107±0.00120±0.06 NE089油用白菜 OilseedCGN06839DH72±0.0049±0.03 NE090油用白菜 OilseedCGN06840DH72±0.7655±1.00 NE091芜菁TurnipUnknownDH122±2.45133±1.98 NE092油用白菜 OilseedL39高代自交系 AIL70±3.6456±4.00 NE094油用白菜 OilseedL33高代自交系 AIL72±0.2846±0.56 NE097油用白菜 Oilseedoil0115高代自交系 AIL77±0.15104±0.22 NE108油用白菜 OilseedL143高代自交系 AIL86±2.47120±2.38 NE109黄籽沙逊油菜Yellow SarsonR-O-18高代自交系 AIL118±0.16101±0.10 续附表1 Continued table S1 编号Code亚群Sub-population品种名称accessions name类型Type试验1Trait 1试验2 Trait 2 开花时间DTF (d)开花时间 DTF (d) NE110芜菁TurnipUnknown高代自交系 AIL115±2.31108±2.55 NE111小白菜Pak ChoiUnknown高代自交系 AIL79±0.0753±1.00 NE112油用白菜 OilseedQian Feng 3地方品种 LV87±0.8298±0.77 NE113油用白菜 OilseedQian Feng 6地方品种 LV101±4.0088±3.90 NE115油用白菜 OilseedXiao Shi Qiao 14地方品种 LV72±3.4660±4.00 NE116油用白菜 OilseedCha He 10地方品种 LV72±1.4157±1.50 NE118油用白菜 OilseedYao An Hei Cai Zi地方品种 LV94±3.6167±4.00 NE119油用白菜 OilseedQi Lu Hei Cai Zi地方品种 LV98±3.10111±3.55 NE121油用白菜 OilseedHua Ning Ai Jiao Hei Cai Zi地方品种 LV74±1.5878±1.98 NE122油用白菜 OilseedHua Ping Ai Jiao Hei Cai Zi地方品种 LV93±5.29NA NE124油用白菜 OilseedWen Shan Ai You Cai地方品种 LV72±2.3264±3.48 NE125油用白菜 OilseedMeng Zi Ai Jiao Hei Cai Zi地方品种 LV72±1.1587±2.28 NE126油用白菜 OilseedJing Dong Bao Dian Ai Jiao You Cai地方品种 LV72±2.8977±3.00 NE127油用白菜 OilseedMeng Zi Shui Tian Xiang Hei Cai Zi 1地方品种 LV72±1.5091±1.05 NE128油用白菜 OilseedMeng Zi Shui Tian Xiang Hei Cai Zi 2地方品种 LV72±3.1057±2.99 NE129油用白菜 OilseedMeng Zi Hei Ai Jiao Cai Zi地方品种 LV76±1.41101±1.50 NE131油用白菜 OilseedZhao Tong Hei Cai Zi地方品种 LV70±1.4156±1.30 NE133油用白菜 OilseedYuan Yang Hei Cai Zi地方品种 LV78±2.5271±3.10 NE134油用白菜 OilseedYu Xi You Cai地方品种 LV80±2.8373±3.00 NE135油用白菜 OilseedMeng Zi Qiao Tou Xiang Hei Cai Zi地方品种 LV88±5.2985±4.99 NE136油用白菜 OilseedZhen Kang Hong You Cai地方品种 LV85±1.7391±1.90 NE139小白菜Pak ChoiUnknown地方品种 LV94±5.35111±5.00 NE140油用白菜 OilseedFeng Yi Hei Cai Zi地方品种 LV93±4.00122±3.60 NE142油用白菜 OilseedJian Chuan Ai Jiao You Cai地方品种 LVNA83±1.55 NE144油用白菜 OilseedMeng Zi Lao Jia Hei Cai Zi地方品种 LV83±3.5481±4.00 NE145油用白菜 OilseedCheng Gong Da Pi Zi 2地方品种 LV81±2.5260±2.50 NE146油用白菜 OilseedHuang Yang Bai You Cai地方品种 LV79±2.8981±3.20 NE149油用白菜 OilseedYong Ping Yan Bei Hei Cai Zi地方品种 LV98±5.0090±4.55 NE150油用白菜 OilseedHe Qing Niu Jie Ai Jiao Zi地方品种 LV100±2.38107±3.00 NE152油用白菜 OilseedYu Xi Yao Ke地方品种 LV90±1.5094±1.71 NE153油用白菜 OilseedLu Shi 1地方品种 LV79±3.5474±3.95 NE154油用白菜 OilseedNan Hua Zhang He Tun Hei Cai Zi地方品种 LV88±8.7271±6.30 NE155油用白菜 OilseedYu Xi Zhou Yong Da Hei Cai Zi地方品种 LV79±5.0068±3.83 NE156油用白菜oilseedHong Cheng Hei Cai Zi地方品种 LV90±2.5282±3.00 NE157油用白菜oilseedZhen Yuan Xin Tang Li Hong You Cai地方品种 LV82±2.38106±3.10 NE158小白菜Pak ChoiUnknown地方品种 LV72±1.1567±1.30 NE159油用白菜oilseedWan Gang Xiang 2地方品种 LV86±3.7076±5.00 NE160油用白菜oilseed Chu Xiong Dong Ping Xiang Hei Cai Zi地方品种 LV72±4.6969±3.50
“NA”表示缺失数据,开花时间为平均数±标准差
“NA” refers to the missing data, Days to flowering (DTF) are means±SD. AIL: advanced inbred line; LV: local variety
附表2 白菜开花时间候选基因的GO注释
Table S2 GO annotation of candidate genes for significant association signals for flowering time in

候选基因Candidate geneGO注释GO annotation Bra023629GO:0000785染色质;细胞组成 Chromatin; Cellular component GO:0003682染色质结合;分子功能 Chromatin binding; Molecular function GO:0005634细胞核;细胞组成 Nucleus; Cellular component GO:0006333染色质组装或解体;生化过程 Chromatin assembly or disassembly; Biological process Bra029347GO:0003700转录因子活性;分子功能 Transcription factor activity; Molecular function GO:0005634细胞核;细胞组成 Nucleus; Cellular component GO:0006355依赖于DNA的转录调控;生化过程 Regulation of transcription, DNA-dependent; Biological process GO:0043565DNA绑定特异序列;分子功能 Sequence-specific DNA binding; Molecular function Bra006322GO:0003700转录因子活性;分子功能 Transcription factor activity; Molecular function GO:0005634细胞核;细胞组成 Nucleus; Cellular component GO:0006355依赖于DNA的转录调控;生化过程 Regulation of transcription, DNA-dependent; Biological process GO:0043565DNA绑定特异序列;分子功能 Sequence-specific DNA binding; Molecular function Bra025655GO:0003677DNA绑定;分子功能 DNA binding; Molecular function GO:0008270锌离子结合;分子功能 Zinc ion binding; Molecular function GO:0045449转录调控;生化过程 Regulation of transcription; Biological process Bra030284GO:0003676核酸结合;分子功能 Nucleic acid binding; Molecular function Bra022192GO:0000155双传感器活性;分子功能 Two-component sensor activity; Molecular function GO:0004673蛋白组氨酸激酶活性;分子功能 Protein histidine kinase activity; Molecular function GO:0004871信号转运活性;分子功能 Signal transducer activity; Molecular function GO:0004872受体活性;分子功能 Receptor activity; Molecular function GO:0005524ATP结合;分子功能 ATP binding; Molecular function GO:0006355依赖于DNA的转录调控;生化过程 Regulation of transcription, DNA-dependent; Biological process GO:0007165信号转换;生化过程 Signal transduction; Biological process GO:0007600感官直觉;生化过程 Sensory perception; Biological process 续附表2 Continued table S2 候选基因Candidate geneGO注释GO annotation GO:0008020G蛋白偶联的光受体活性;分子功能 G-protein coupled photoreceptor activity; Molecular function GO:0016020生物膜;细胞组成 Membrane; Cellular component GO:0018106肽组氨酸磷酸化;生化过程 Peptidyl-histidine phosphorylation; Biological process GO:0018298蛋白发色团联动;生化过程 Protein-chromophore linkage; Biological process GO:0045449转录调控;生化过程 Regulation of transcription; Biological process Bra024329GO:0005525GTP结合;分子功能 GTP binding; Molecular function GO:0005622细胞内的;细胞组成 Intracellular; Cellular component GO:0007264小GTP酶介导的信号转导;生化过程 Small GTPase mediated signal transduction; Biological process GO:0015031蛋白运输;生化过程 Protein transport; Biological process Bra017569GO:0006511泛素依赖的蛋白讲解过程;生化过程 Ubiquitin-dependent protein catabolic process; Biological process GO:0031461cullin-RING泛素连接酶复合体;细胞组成 Cullin-RING ubiquitin ligase complex; Cellular component GO:0031625范素蛋白连接酶绑定;分子功能Ubiquitin protein ligase binding; Molecular function Bra009510GO:0003700转录因子活性;分子功能Transcription factor activity; Molecular function GO:0005634细胞核;细胞组成Nucleus; Cellular component GO:0006355依赖于DNA的转录调控;生化过程Regulation of transcription, DNA-dependent; Biological process GO:0030528转录调控活性;分子功能Transcription regulator activity; Molecular function GO:0043565DNA绑定特异序列;分子功能Sequence-specific DNA binding; Molecular function
Genome-Wide Association Studies for Flowering Time in
GAO BaoZhen1, LIU Bo1, LI ShiKai2, LIANG JianLi1, CHENG Feng1, WANG XiaoWu1, WU Jian1
(1Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences, Beijing 100081;2Institute of horticultural crops, Yunnan Academy of Agricultural Sciences, Kunming 650205)
【Objective】To identify the genetic loci or candidate genes for flowering time regulation infor improvement of pre-mature bolting resistance of. 【Method】In this study, 116germplasm accessions were selected to evaluate flowering time variations in greenhouse and open-field, respectively. Total genomic DNA was extracted with 1.2x re-sequenced depth. filtering, mapping with reference by Pooled Mapping was conducted to obtain a genomic high quality SNP set. Then the population structure and linkage disequilibrium (LD) were analyzed using SNP set after condition filtering. In total 2000 SNP points were selected from all SNPs randomly to conduct phylogenetic tree analysis using PhyML software with maximum likelihood method. all high quality SNPs were used to conduct genomic linkage disequilibrium analysis with Haploview software. Genome-wide association study (GWAS) for flowering time variations was then conducted based on software TASSEL, GAPIT and R. According to the position of strong association signals and LD block, the candidate signals for flowering time were identified. Eventually, flowering time candidate genes inwere predicted by gene colinearity relationship betweenand, and gene function annotation.【Result】The 116accessions showed extensive variations in flowering time. Significant variation was also observed between greenhouse and open-field environments. The distribution of flowering time under open-field was partial normal, while the flowering time distributed evenly under greenhouse. Phenotypes of flowering time were significantly correlated between different environments, indicating that genetic effect played a crucial role in regulation of flowering time. A total of 1.03 million SNPs covering genome-wide were generated by biotechnology analysis. Population structure showed that accessions from each sub-group were clustered, and had a close relationship with geographic origin in phylogenetic tree. The linkage disequilibrium decay across genome-wide was 2.3 kb, demonstrating that there were frequent recombinations and variants in 116accessions. A total of 54 strong signals (>4) were detected using mixed linear model and 87 (>5) using general linear model under two different environments. Thirty-three strong signals (27 loci under greenhouse, 19 loci under open-field) were saved after considering LD block (2>0.33), including 13 co-identified SNPs. Based on genome colinearity betweenand, and gene function annotation, 14 candidate genes were predicted. Three candidate genes,,,, were co-identified under greenhouse and open-field environments., a key gene involved in flowering time regulation was also identified under open-field condition.【Conclusion】Correlation analysis of flowering time under different environments indicated that genetic control is a decisive effect on flowering time. A total of 33 significant associated SNPs controlling flowering time were identified by GWAS. By combining LD block, genome colinearity betweenand, and gene annotation, 14 flowering time candidate genes were predicted.
; flowering time; linkage disequilibrium; genome-wide association study; candidate gene
2017-01-20;接受日期:2017-03-15
国家自然科学基金(31272179)
高宝祯,E-mail:18853812686@163.com。通信作者武剑,Tel:010-82105971;E-mail:wujian@caas.cn
猜你喜欢
作物研究(2020年5期)2020-12-08
科学导报(2020年26期)2020-06-09
新农业(2019年20期)2019-11-02
科学导报(2019年67期)2019-05-08
北京农学院学报(2019年1期)2019-02-22
现代园艺(2018年2期)2018-03-15
作文世界(小学版)(2017年8期)2017-09-07
现代园艺(2017年11期)2017-06-28
河南农业科学(2017年4期)2017-04-12
作文大王·低年级(2017年1期)2017-02-16
