
匍匐翦股颖接种立枯丝核菌后基因表达变化的转录组学分析
2017-09-26 08:02史毅牛奎举马晖玲
中国农业科学 2017年17期
史毅,牛奎举,马晖玲
匍匐翦股颖接种立枯丝核菌后基因表达变化的转录组学分析
史毅,牛奎举,马晖玲
(甘肃农业大学草业学院/草业生态系统教育部重点实验室/中-美草地畜牧业可持续发展研究中心,兰州 730070)
【目的】确定匍匐翦股颖()接种立枯丝核菌()后基因种类和表达量在转录水平的变化规律,明确草坪草病原菌侵染响应的关键基因。【方法】匍匐翦股颖生长14 d后采用麦粒培养物接种立枯丝核菌,接种3 d后选取感病叶片和未接种叶片提取RNA,进行转录组高通量测序,然后利用生物信息学分析,用Trinity组装匍匐翦股颖转录组,以组装子为参考,以|log2(fold change)|>1,-value<0.005为阈值选取感病和健康匍匐翦股颖叶片转录组的差异表达基因,并用iTAK软件分析其转录因子家族及表达变化,与植物R基因库进行blast分析R蛋白分类、利用Mapman软件分析生物胁迫信号通路相关基因的表达变化。【结果】高通量测序得到125 253 092条高质量待分析reads。经Trinity从头组装后,得到466 761条转录本。过半数的转录本长度为700 bp以上,组装结果N50=1 100 bp。使用CD-HIT选择334 212条转录本(所有转录本的71.60%)作为Unigene,平均长度573 bp,N50=791 bp。接种后植物比接种前植物基因有7 937个上调表达,1 570个下调表达。上调基因中296个,下调基因有142个都可被定义为转录因子,分布在58个转录因子家族中,其中锌指蛋白包含转录因子C2H2最多,有54个,C3H次之,为22个。差异基因中451个可定义为植物R蛋白表达基因,可分为33类,其中包含NBS-LRR结构域的抗病蛋白、LRR受体蛋白激酶、ABC-2类型的转运蛋白、U-box结构域蛋白激酶和热激蛋白这5类基因变化最显著。差异基因中大量上调表达基因可富集在病原识别、活性氧消除、信号传导、细胞凋亡、病程相关蛋白等生物胁迫相关基因类别,下调基因显著富集在植物生长发育相关类别和通路。qRT-PCR验证了随机挑选差异基因的表达量变化,均与RNA-seq分析结果一致,其中包括12个C2H2转录因子基因,10个C3H转录因子基因,以及12个R蛋白基因。【结论】病原菌侵染后,引起匍匐翦股颖大量基因表达变化,其中转录因子、R蛋白以及抗性相关基因多为上调表达,作用为抑制病原菌扩散,而生长发育相关基因表达下调,这些进程共同使匍匐翦股颖产生对立枯丝核菌的先天基础抗性。
匍匐翦股颖;立枯丝核菌;转录组;差异表达基因;抗病机制
0 引言
【研究意义】匍匐翦股颖()作为草坪草应用广泛[1],其不定根入土较浅,喜水,但潮湿的生长环境使其易感病[2-3]。草坪褐斑病是所有草坪病害中分布最广的病害之一,是中国冷季型高尔夫草坪果岭上发生最普遍、最严重的病害之一[4],该病常发于夏季,其病原能在极短的时间内迅速毁灭草坪,防治存在一定困难[5]。明确草坪草对褐斑病的抵抗机制,对于更好地解决高尔夫草坪病害具有重要指导意义。【前人研究进展】褐斑病最早于1914年在草坪草上被发现,后于1919年由Piper等[6]首次报道其病原菌为立枯丝核菌(),之后又被众多的研究证实[7-9]。立枯丝核菌最适生长温度为30℃,此时生长速度最快,所以草坪褐斑病一般在温暖潮湿的区域发病率较高,而在寒温带则多发于夏季,该菌是一种土壤习居菌,传播迅速,寄主范围广,几乎可以侵染所有草坪草,冷、暖季型禾本科草坪草上均有报道该病害的发生及其危害性[10-11],条件适宜时,发病速度快,影响范围广,破坏性大,成为草坪建植与管护的主要限制因子[5]。国内外学者从生理生化角度对多种植物的抗病性及其信号转导机制进行了研究,并有了较清晰的认识,植物受到病原菌侵害后,产生R蛋白(resistance protein)用以识别病原菌效应蛋白,植物R蛋白分为很多类,其中NBS-LRR蛋白是R蛋白中最大的一个分类,在植物被病原菌侵染过程中可直接或间接地识别特定的病原效应因子[12],并且一部分NBS-LRR基因可直接作用于病原菌分泌产生的效应因子,抑制植物体因病菌的效应因子作用而提高的敏感性[13]。然后植物产生小分子抑菌物质,如酚类化合物和植保素等,以及各种病程相关蛋白(pathogenesis related proteins,PR蛋白)等,抑制病菌繁殖,同时启动自身超敏反应,导致自体细胞凋亡,阻止病原菌进一步扩散[14]。在这整个过程中,基因的大量表达是靠转录因子调节,转录因子在植物抗生物及非生物胁迫过程中的变化也是近年来的研究热点。【本研究切入点】常规植物抗病机制研究多针对某种植物数个基因或蛋白,采用基因定量或蛋白定量的方法,研究单一通路,基因或蛋白数量越多耗费人力财力和时间越多,并且植物抗病是一个网络式的信号变化,单一物质或通路不能全面说明问题。随着基因测序技术飞速发展,高通量转录组测序已成为非常普遍的基因网络变化的研究方法,因其数据通量高,覆盖全面,用时短,操作方便,多被研究者用于大量信号转导的机理研究,将其用于植物抗病机制,可全局性把握植物抗病信号传导中涉及到的大量基因变化。目前,转录组学用于植物和病原菌互作的研究较多[15-16],但用转录组学方法进行立枯丝核菌和匍匐翦股颖互作的研究尚未见报道。【拟解决的关键问题】利用大规模数据分析技术对匍匐翦股颖被立枯丝核菌侵染前后的基因变化进行全局分析,找到草坪草病原菌侵染响应的关键基因,为匍匐翦股颖的抗病育种提供理论依据和数据信息。
1 材料与方法
试验于2015—2016年在甘肃农业大学完成。
1.1 供试材料
供试植物:匍匐翦股颖商用品种‘PennA4’由北京克劳沃公司提供;供试菌株:立枯丝核菌购自中国科学院菌种保存中心。
1.2 制备方法
1.2.1 植物材料制备 匍匐翦股颖种子经浸泡消毒后自然风干。以灭菌的沙土混合物(1﹕2)为基质,2015年9月种于240 ml组培瓶中,每瓶约0.25 g种子均匀撒入,封口膜遮盖,于甘肃农业大学草业学院恒温培养室中培养,温度为20℃昼/18℃夜,光周期为16 h光照/8 h黑暗,2周后接种病原菌。
1.2.2 病原菌接种 采用Cortes-Barco等[17]的接菌物制备方法,利用麦粒培养基制备接菌物,并将其打碎研磨成粉末。接菌时,先从瓶口向内喷水雾,使植物叶片湿润,然后向叶片撒播接菌物粉末,每瓶0.1 g,使其均匀分布在瓶内植物的叶片上。封口膜遮盖培养于恒温培养室,培养条件同前,每天观察病情。
1.3 测序样品准备和RNA提取
植物材料接菌前、接菌3 d后分别取样,各4个重复,每瓶内所有叶片为一个样,剪取叶片用锡箔纸包裹后迅速置于液氮中低温保存。所有样品用植物RNA提取试剂盒(OMEGA)提取RNA,具体方法见说明书。经琼脂糖凝胶电泳鉴定,测量浓度,RIN(RNA integrity number)指数后选择完整性好,纯度浓度高,RIN>6.3的RNA样品用于建库测序。
1.4 建库和测序
接菌前和接菌后3 d的匍匐翦股颖叶片RNA样品,分别由4个重复样品混合而成,用于构建cDNA库。测序使用Illumina HiSeq平台,由诺禾致源公司(北京)完成。
1.5 转录组组装
测序得到的原始数据(raw reads)使用立枯丝核菌基因组(Genome ID 11579)去除污染序列之后再进行后续步骤。使用FASTQC确定待分析数据(clean reads)质量,Trinity[18](https://github.com/trinityrnaseq/ trinityrnaseq/wiki)用于转录本的从头组装(assembly),然后使用CD-HIT(http://weizhongli-lab. org/cd-hit/)选取相似类中最长的作为唯一基因,得到所有基因序列集合(Unigene)作为分析基因表达量的参考序列(reference sequences)。
1.6 差异表达基因(differential expression genes,DEGs)数字分析及荧光定量PCR分析
用测序得到的各库clean reads映射(map)于组装得到的参考转录组,然后使用DEGseq 对各Unigene在各库的reads数量(均一化后)进行分析,以|log2(fold change)| >1,-value (adjusted-value)<0.005[19]为阈值选取差异表达基因。
选取病原菌响应植物基因34个,引物设计见表1,使用SYBR Green染料进行荧光定量PCR,利用DDCT法得到各基因在各植物样本中的表达量。
20 ml反应体系:SYBR 10 ml,上下游引物各0.8 ml,模板5 ml,去离子水3.4 ml。反应条件:95℃ 10 min;40 cycles:95℃ 15 s,57℃ 1 min。
1.7 基因功能注释及通路富集
Unigene和差异基因的功能注释均使用相应软件与基因功能各大数据库进行比对,以e-value<10-5为阈值来确定可信的基因功能。
1.8 数据统计分析
差异基因表达量统计分析使用SAS软件T-test方法(<0.05)。
2 结果
2.1 转录组组装与注释
利用Illumina 2×150 bp双端测序共测得128 539 186条reads,产生18.7 G数据,去除接头和低质量序列后,得到125 253 092条高质量待分析reads。经Trinity从头组装后,得到466 761条转录本(表2)。过半数的转录本长度为700 bp以上,组装结果N50=1 100bp。使用CD-HIT选择334 212条转录本(所有转录本的71.60%)作为Unigene,平均长度573 bp,N50=791 bp。转录本和Unigene长度分布见图1-A。

表1 qRT-PCR验证基因选择及引物设计
用334 212条Unigene与基因功能注释数据库的比对结果显示(表2),130 367(39%)条序列在NCBI的非冗余蛋白序列数据库(NR)中可注释到,72 777(21.77%)条可注释到NCBI的非冗余核酸数据库(NT),108 363(32.42%)条可注释到基因本体论(GO)数据库。与NR数据库的注释结果显示,与匍匐翦股颖相似度最高的物种是粗山羊草(),同源基因达到Unigene总数的13%(17 166),其次同源率较高的为二穗短柄草()和大麦(),同源序列分别为15 384条(12%)和12 167条(9%),另外,26 127条(20%)Unigene与其他禾本科重要物种相似,包括乌拉尔图小麦()、水稻()、玉米()和高粱()等。同源物种比对结果见图1-B。

表2 匍匐翦股颖转录组测序、组装统计以及功能注释结果统计
2.2 差异基因数量
使用DEGseq对各Unigene在各库中map到的read数量(均一化后)进行分析,以|log2(fold change)| >1,-value (adjusted-value) <0.005为阈值,得到7 937个上调表达基因,1 570个下调表达基因,如图2所示,log2(fold change)在-1到1之间而不为0的也是差异基因,但其差异倍数未达到阈值,所以不计算在差异基因内。接菌后植物基因表达上调明显多于下调。
2.3 差异基因中转录因子分类
从所有差异基因挑出转录因子相关基因,上调表达基因中有296个,下调表达基因有142个可被定义为转录因子表达基因。如表3所示,差异基因分布在58个转录因子基因家族中,其中不乏与植物抗逆密切相关的转录因子家族。54个上调基因为C2H2转录因子基因家族,丰度最高,其次为22个上调基因定义为C3H转录因子基因家族。GNAT、SNF2、Zn-clus和bZIP家族也有较多差异基因可注释到。下调基因中,AP2-EREBP和AUX/IAA生长素相关转录因子较多,这与KEGG通路富集结果相符,较多的下调基因富集在植物激素信号传导通路上。其中orphans为尚未确定功能及分类的转录因子。

表3 匍匐翦股颖差异基因中转录因子分类
选取其中差异基因最多的两类,C2H2转录因子12个(图3-A),C3H转录因子10个(图3-B)进行qRT-PCR验证,基因表达趋势均与RNA-seq分析相同,且大部分qRT-PCR检测表达量变化均大于RNA-seq分析同期表达量,表明这些基因的实际表达变化趋势和RNA-seq结果相符,同时也说明了RNA-seq结果可信。
2.4 差异基因中R基因的变化
植物R基因数据库(plant resistance gene database,PRGDB)是一个开放性的、每日更新的、包含现被发现的所有植物R家族基因的数据库[20](http://prgdb. crg.eu/wiki/Main_Page)。将匍匐翦股颖9 507个差异基因与该数据库进行比对,451个基因可比对到相应的不同结构的R基因,可分为33类(图4),其中未确定特定结构和功能的R基因最多,其次为包含核酸结合位点-亮氨酸富集(NBS-LRR)结构域的R基因、亮氨酸富集类受体蛋白激酶(LRR-RLK)、ABC-2类型的转运蛋白、包含U-box结构域的蛋白激酶以及热激蛋白等。将此5类结构功能明确的基因挑出,观察其表达量变化,结果表明此5类R基因均有超过半数为上调表达,最高上调倍数达14.72倍,其中热激蛋白基因几乎全部上调表达(图5-A)。随机挑选12个基因,使用qRT-PCR检测其接菌前及接菌后的表达量变化,12个基因的变化趋势均与RNA-seq分析结果相符,并且与转录因子相似,大部分qRT-PCR检测表达量高于RNA-seq分析表达量(图5-B)。
2.5 差异基因的相关通路分布
9 507个差异基因在有关Pathway的主要公共数据库KEGG[21]的显著富集结果见表4,可看出上调基因主要富集在核糖体蛋白相关通路、MAPK信号通路、吞噬体形成、赖氨酸合成、蛋白酶体形成和细胞自噬作用等通路上。下调基因主要富集在植物激素信号传导、mRNA形成过程中的剪接小体合成、固醇类和苯甲酸合成、半乳糖代谢、光合作用以及生理节奏调节等通路。

X轴代表接菌后基因表达比接菌前基因表达的变化倍数;Y轴代表基因表达量变化的统计学显著程度,-lg(校正后的p-value)越大,即差异越显著,蓝色部分代表无显著差异的基因,红色部分表示有显著性差异的上调基因,绿色部分表示显著差异的下调基因

A:12个随机挑选C2H2转录因子表达量The expression level of 12 random-selected C2H2 TFs;B:10个随机挑选C3H转录因子表达量The expressionlevel of 10 random-selected C3H TFs
利用Mapman软件将差异基因功能和表达图形化,在整个生物胁迫相关的信号传导中,首先变化的是由病原菌引起寄主呼吸爆发同时引起的氧化还原相关基因(redox state),以及谷胱甘肽转移酶(glutathione-S-transferase)基因,均为较大规模的上调表达,过氧化物酶有较低程度的上调表达。在接下来的信号传导过程中,大规模的基因上调表达,乙烯(ethylene)和茉莉酸(JA)作为激素信号,也有较大程度的上调,而其他激素并未见明显的上调表达。在各种水解酶类中,2个-葡聚糖酶基因上调表达,与细胞壁相关酶类和蛋白水解(proteolysis)相关酶均有大批基因,且大部分成上调表达。有趣的是,病程相关蛋白(PR proteins)并没有较多基因上调(图6)。与PRG数据库注释结果一致,热激蛋白大部分基因上调表达。与植物病原响应相关的次级代谢产物合成基因也呈上调表达。

1:未确定功能Undetermined function;2:包含NBS-LRR结构域的抗病蛋白NBS-LRR domains-containing disease resistance protein;3:亮氨酸富集的类受体蛋白激酶Leucine-rich repeat receptor-like protein kinase;4:ABC-2型转运子蛋白家族ABC-2 type transporter family protein;5:包含U-box的蛋白激酶U-box domain-containing protein kinase;6:热激蛋白70 Heat shock cognate protein 70;7:DEAD/DEAH盒子螺旋酶DEAD/DEAH box helicase;8:AP2结构域类转录因子AP2 domain class transcription factor;9:吡哆醛磷酸盐依赖型转移酶超家族Pyridoxal phosphate (PLP)-dependent transferases superfamily protein;10:黄素单加氧酶蛋白家族Flavin-binding monooxygenase family protein;11:a型输入蛋白Importin alpha isoform;12:核糖体蛋白Ribosomal protein;13:S结构域2, 5表达蛋白S-domain-2, 5 expressed protein;14:网格蛋白重型链Clathrin heavy chain;15:细胞分裂控制蛋白Cell division control protein;16:染色体压缩调节蛋白家族Regulator of chromosome condensation (RCC1) family protein;17:七跨膜MLO蛋白家族Seven transmembrane MLO family protein;18:WRKY转录因子WRKY transcription factor;19:蔗糖合成酶mRNA Sucrose synthase mRNA;20:包含NAC结构域蛋白NAC domain containing protein;21:泛素E3连接酶mRNA Ubiquitin E3 ligase mRNA;22:TTF型锌指蛋白包含HAT二聚体结构域TTF-type zinc finger protein with HAT dimerisation domain;23:ATRFC1型复制因子ATRFC1 replication factor;24:类RPP1型抗病蛋白RPP1-like disease resistance protein;25:拟南芥结构分子Arabidopsis thaliana structural molecule;26:囊泡相关蛋白Vesicle-associated protein;27:GATA转录因子GATA transcription factor;28:Lon蛋白酶Lon protease;29:腺苷酸环化酶Adenylate cyclase;30:渗调蛋白Osmotin;31:黄烷酮醇-4-还原酶Dihydroflavonol-4-reductase;32:类RH8A型抗病蛋白RPH8A-like disease resistance protein;33:RNA聚合酶II RNA polymerase II
3 讨论
匍匐翦股颖PennA4接种病原菌立枯丝核菌3 d后,病原菌诱导或抑制大量基因表达变化,其中上调表达基因数量和幅度均多于下调表达基因,并且有58个转录因子家族基因差异表达,说明众多基因表达受到调控。诱导上调的转录因子中,C2H2转录因子最多,C3H次之。C2H2和C3H转录因子是主要的锌指蛋白结构域包含转录因子。Zhang等[22]使用病毒诱导基因沉默系统(VIGS),发现核盘菌SScut蛋白激发子引起的烟草免疫反应过程中,C2H2锌指蛋白(NbCZF1)是一个重要的调节因子,NbCZF1沉默的植株很大程度地抑制了SScut诱导的烟草免疫反应过程。棉花的一个C3H转录因子GhZFP1从盐胁迫诱导的棉花植株中克隆出来,使其在烟草中过表达,发现过表达GhZFP1的烟草不但提高了对盐胁迫的耐受能力,并且产生了较高的对病原菌的抗性[23]。上述研究说明C2H2、C3H等锌指蛋白转录因子在植物抗病过程中的重要性,与本研究中匍匐翦股颖在立枯丝核菌侵染后锌指蛋白转录因子基因大量上调表达的结果互相印证。BZIP、AP2-ERF、WRKY和MYB转录因子是最多报道与植物抗病相关的转录因子,Gao等[24]从基因组水平分析了玉米在接种后的基因表达变化,发现B3、AP2、WRKY转录因子上调表达;Venu等[25]利用基因芯片研究水稻转录组变化,发现在侵染后,除了C2H2转录因子上调之外,MYB、WRKY、bZIP和NAC等转录因子基因也有大量上调或特异表达;Wang等[26]研究水稻接种后转录组变化,同样发现MYB和WRKY转录因子起主导作用。在本研究中,与上述研究类似,MYB转录因子有10个上调表达,bZIP转录因子有12个上调表达,但不同的是,有7个MYB转录因子和10个bZIP转录因子同时下调表达,而AP2-ERF和WRKY转录因子几乎没有上调表达,只有一部分的下调,这种差异可能由于匍匐翦股颖和水稻、玉米虽同为禾本科,却在遗传结构和生长习性上有很大差异,而菌种不同也导致很大区别,即使同为,其也有很多不同株系。而在Yan等[27]的报道中,WRKY转录因子在烟草中过表达却引起了转基因烟草对抗性的下降。所以说在不同物种,不同菌株,不同基因家族,甚至同一基因家族内部的不同基因,其变化趋势也有所差异。
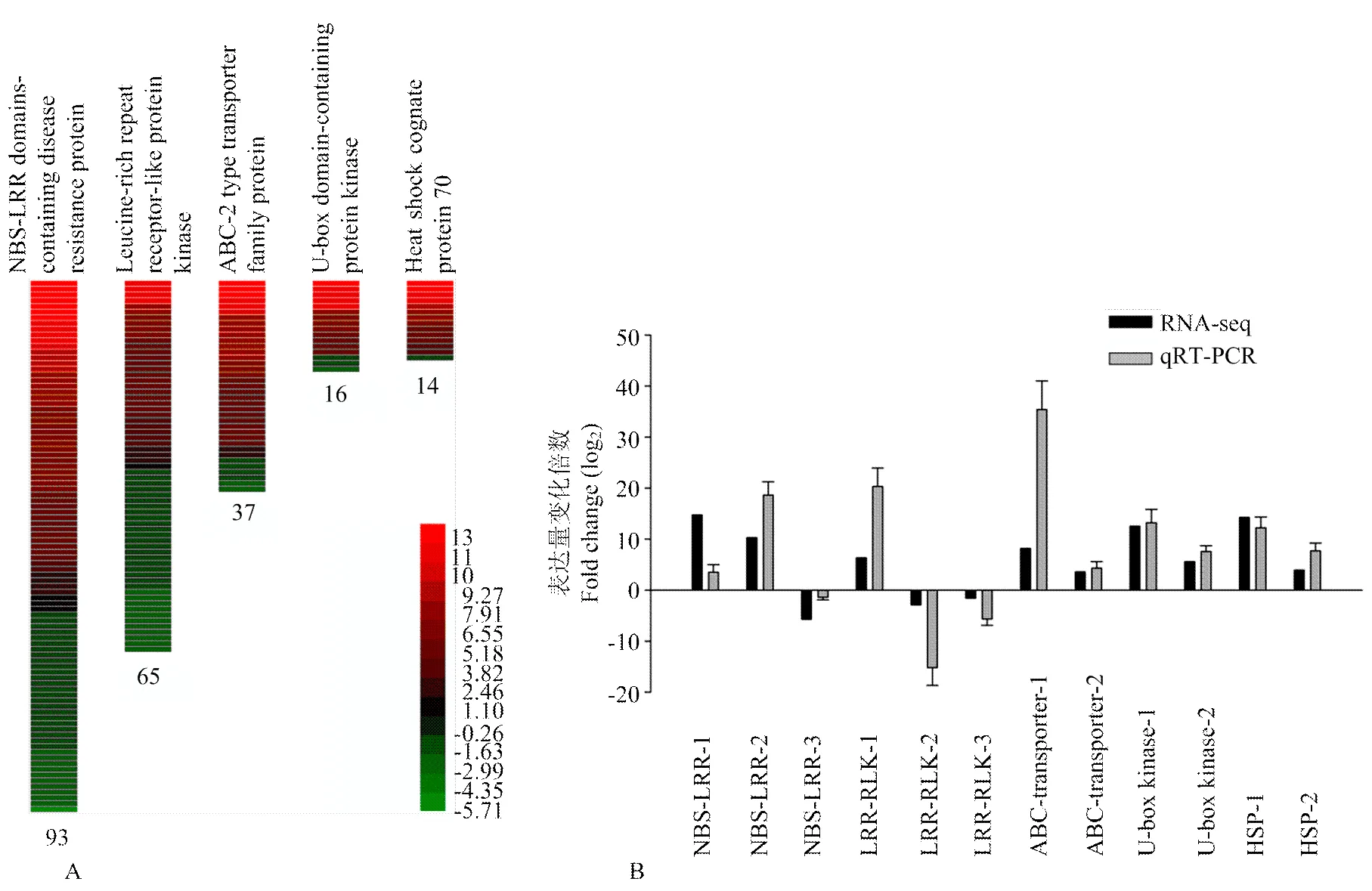
A:变化基因数目最多的前5类基因RNA-seq分析表达量变化,一个长方框代表一个基因,绿色代表下调表达,红色代表上调表达,变化倍数见图例The gene expression changes of the top 5 R-gene families analyzed by RNA-seq. Each small bar presented one gene. Red indicated up-regulated while green indicated down-regulated. The fold change value could be identified by the figure legend;B:qRT-PCR分析随机挑选的此5类R基因表达量,共挑选12个The expression change of random selected gene from 5 R-gene families, 12 genes were selected
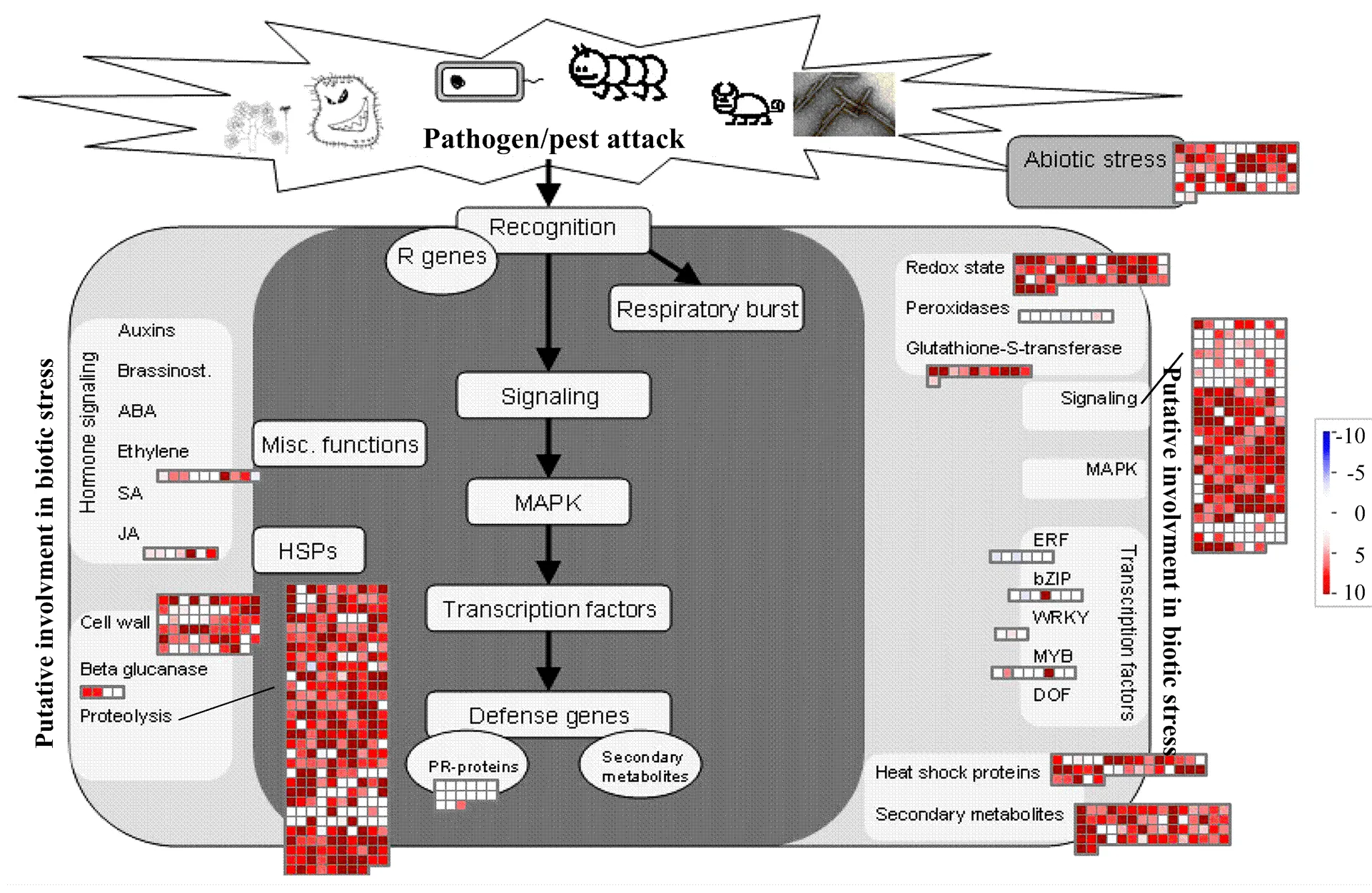
图6 匍匐翦股颖差异基因在植物生物胁迫相应通路中的分布

表4 匍匐翦股颖差异表达基因KEGG富集通路
通过与植物R基因数据库比对,发现33类共451个R基因被病原菌诱导,表达量有较大变化。其中数量最多且结构功能明确的是包含核酸结合位点-亮氨酸富集(NBS-LRR)结构域的R基因。Yang等[28]从玉米、高粱、二穗短柄草克隆得到8个NBS-LRR基因,将其转入水稻,发现其中6个基因的转化植株对1—3个水稻稻瘟病原有抗性,说明NBS-LRR基因对植物抗病机制的调节作用,这与本研究结果植物在病原菌侵染后NBS-LRR基因作为与抗性相关基因上调表达一致。本研究中,匍匐翦股颖差异表达基因中的R基因还包括亮氨酸富集类受体蛋白激酶,作为细胞表面信号分子受体的第二大分类,LRR-RLK在感受植物病原信号和病原菌识别中起重要作用[29],同时也参与植物抗逆响应[30],Godiard等[31]报道ERECTA,一个LRR-RLK蛋白是控制拟南芥产生对细菌萎蔫抗性的重要蛋白,Parrott等[32]也报道过一个LRR-RLK蛋白是大麦的真菌病害识别蛋白,对大麦抵抗真菌病害有重要作用。但也有过报道,在拟南芥中BIR2作为一个LRR-RLK,可以限制另一细胞表面识别激酶BAK1,从而反向调节PAMP(病原相关分子模式)触发的植物免疫机制[33]。可以看出LRR-RLK参与植物抗逆响应在不同植物中有不同的方式。植物R基因ABC-2类型的转运蛋白,包含U-box结构域的蛋白激酶以及热激蛋白激酶同时可在差异基因中找到,其中ABC-转运蛋白全称为ATP结合盒转运蛋白,它参与植物体内众多信号的传导,包括与植物抗逆相关的激素信号、外界毒素、具有抗菌作用的次生代谢产物等[34-35]。U-box蛋白大部分是泛素系统中决定底物识别特性的泛素连接酶,它参与植物的先天免疫反应,例如病原物识别、抗性调节信号传导等[36]。González-Lamothe等[37]通过在烟草中过表达Ubox结构域包含蛋白CMPG1以及沉默番茄等一系列实验,证明是植物防御及抗病必不可少的基因。本研究中匍匐翦股颖U-box蛋白在立枯丝核菌侵染后大量上调,在此得到了证明。但也有报道证明在受冷后过表达U-box蛋白基因VaPUB的葡萄植株,接种后,病程相关蛋白PR10的表达量显著下调[38]。说明在不同物种中,蛋白和基因的表达调节差异显著。热激蛋白是逆境响应蛋白,包括生物和非生物胁迫逆境[39],它常结合于多种抗逆基因的启动子,来调节该基因的表达。以上匍匐翦股颖R基因均为病原菌响应基因,大多有识别病原物及其效应因子的功能,一部分有调节抗逆基因表达的功能。
植物为了减少活性氧对自身的伤害,需要进行一系列的氧化还原反应来调节细胞内活性氧的含量[40],在图6中体现为氧化还原态相关基因的大量上调。谷胱甘肽还原酶也起到清除活性氧的作用[41]。Liebrand等[42]研究表明在植物自身识别受体识别病菌分子,并产生R基因识别病原菌效应蛋白的过程中,需大量的信号传导来诱发植物自身超敏反应,这与本研究差异基因KEGG通路富集结果(表4)相符,MAPK信号通路有大量基因富集,在较近的对MAPK信号通路的综述中提到[43],在水稻中有6个MAPK基因被鉴定出,其中4个被认为与植物抗性相关。MAPK信号通路参与多种植物抗性响应,包括植物抗性激素的合成、ROS产生、气孔关闭、抗性基因表达、细胞壁强度改变、细胞凋亡等[43]。植物激素的产生和转移可调节植物在病菌侵染过程中的生长,同时也是信号传导分子,来帮助植物识别病原菌并产生一系列反应,其中茉莉酸和乙烯是重要的信号激素[44],匍匐翦股颖在立枯丝核菌侵染后,激素信号相关基因也有高倍上调,与此结论相符。Bellincampi等[45]认为植物细胞壁是一个动态变化的结构,遇到不同的环境胁迫以及病原菌侵染过程,其结构都会产生不同的动态变化,病原菌不只要从寄主细胞获得营养,对其细胞壁产生破环,还要保持一部分寄主细胞的活性,使菌自身可以依存并繁殖。这与本研究中匍匐翦股颖被病原菌诱导上调的基因中有大量与细胞壁有关相类似。被侵染后,植物体针对自身侵染部位也有相应的调节机制,KEGG通路富集结果表明吞噬体形成、蛋白酶体形成和细胞自噬作用等通路有大量基因富集,说明匍匐翦股颖已启动自体细胞凋亡机制。与植物体抗性产生相应的是,植物正常生长受到破坏,所以其代谢功能、光合作用、生理调节功能相关基因均下调表达,下调基因在KEGG通路富集结果可印证这一点。综上所述,通过分析匍匐翦股颖病菌诱导差异表达基因在生物胁迫相关通路中的分布和变化,可以看出差异基因基本与通路相符合,可以看到匍匐翦股颖在立枯丝核菌侵染后的一个完整的响应通路。
在对植物接种病原菌后转录水平基因变化的研究中,研究较多的植物是水稻,MatiĆ等[46]研究不同敏感度的水稻基因型接种水稻恶苗病菌后基因表达变化,其在植物病原响应通路中差异基因的分布与本研究不同的是,敏感品种有大量PR蛋白上调表达,抗性品种却有大量PR蛋白下调表达,而本研究结果中PR蛋白基本无显著变化,产生这种不同的原因可能是,为种内传播的病菌,而为土壤传播的病菌,接种方式、病菌生长方式均不同,并且PennA4为中度敏感的匍匐翦股颖品种,说明植物抵抗病菌的方式因病菌种类、植物敏感程度的不同也会有差别。匍匐翦股颖与病原菌互作的转录组学研究较少,2012年,Orshinsky等[47]利用转录组学研究了匍匐翦股颖与币斑病菌()互作的基因变化,其接菌4 d的植物抗性相关基因变化较少,NBS-LRR基因仅有3个产生差异,ABC-转运蛋白仅6个基因变化,蛋白水解酶类仅1个变化,而其与本研究结果差别如此之大的原因应为当时用于转录组测序的为GS-FLX 454测序平台,较陈旧,数据量少,故分析得到的差异基因数量少很多。
4 结论
立枯丝核菌的侵染引起匍匐翦股颖58个转录因子家族表达变化,其中C2H2和C3H锌脂蛋白转录因子家族差异基因最多,分别为54个和22个。451个差异表达基因可定义为植物R基因,分为33类,其中差异基因数量最多的5类为NBS-LRR抗病基因、LRR受体蛋白激酶、ABC-2类型的转运蛋白、U-box结构域蛋白激酶和热激蛋白。同时差异表达基因可富集在病原识别、活性氧消除、信号传导、细胞凋亡、病程相关蛋白等生物胁迫相关基因类别。qRT-PCR验证了34个差异基因的表达量变化,均与RNA-seq分析结果一致。
References
[1] 周胜, 陈本建, 周玉雷, 赵琛, 蓝祖庆, 陈平. 匍匐翦股颖新品系坪用性状研究. 中国农学通报, 2008, 24(5): 181-185.
ZHOU S, CHEN B J, ZHOU Y L, ZHAO C, LAN Z Q, CHEN P. Study on turf trait ofNo.2 radiation new strain., 2008, 24(5): 181-185. (in Chinese)
[2] 孙吉雄. 草坪学. 2版. 北京: 中国农业出版社, 2003.
SUN J X.. Beijing: China Agriculture Press, 2003. (in Chinese)
[3] BENELLI J J, HORVATH B J, BROSNAN J T, KOPSELL D A. Plant health characteristics of creeping bentgrass affected by strobilurin fungicide applications and turfgrass diseases., 2016, 56(2): 862-869.
[4] 殷萍萍, 李珊珊, 王玉玲, 曾会明. 立枯丝核菌 () 对日本结缕草 (steud.) 的侵染过程研究. 微生物学通报, 2015, 42(7): 1253-1262.
YIN P P, Li S S, WANG Y L, ZENG H M. The infection process ofin,2015, 42(7): 1253-1262. (in Chinese)
[5] 马晖玲, 房媛媛. 植物抗病性及诱导抗性在匍匐翦股颖病害防治中的应用. 草业学报, 2014, 23(5): 312-320.
MA H L, FANG Y Y. Induction of plant disease resistance and its application for disease control in creeping bentgrass., 2014, 23(5): 312-320. (in Chinese)
[6] PIPER C V, COE H S. Rhizoctonia in lawns and pastures., 1919, 9: 89-95.
[7] RAIKES C, BURPEE L L. Use of multispectral radiometry for assessment of Rhizoctonia blight in creeping bentgrass., 1998, 88(5): 446-449.
[8] QIN Y, MILLER C J, WHITE J F, RICHARDSON M D. Isolation and characterization of fungal inhibitors from., 2000, 48(10): 4687-4692.
[9] BLOK W J, LAMERS J G, TERMORSHUIZEN A J, BOLLEN G J. Control of soilborne plant pathogens by incorporating fresh organic amendments followed by tarping., 2000, 90(3): 253-259.
[10] 余德亿, 翁启勇, 汤葆莎. 几种冷季型草坪草病害及病原种类. 草业科学, 2003, 20(1): 59-64.
YU D Y, WENG Q Y, TANG B S. Diseases and pathogens reported on 4 cool-season turfgrasses., 2003, 20(1): 59-64. (in Chinese)
[11] 余德亿, 汤葆莎, 翁启勇. 几种暖季型草坪草病害及病原种类. 草业科学, 2001, 18(4): 60-64.
YU D Y, TANG B S, WENG Q Y. Diseases and pathogens reported on 4 warm-season turfgrasses., 2001, 18(4): 60-64. (in Chinese)
[12] DEYOUNG B J, INNES R W. Plant NBS-LRR proteins in pathogen sensing and host defense., 2006, 7(12): 1243-1249.
[13] MEYERS B C, KOZIK A, GRIEGO A, KUANG H, MICHELMORE R W. Genome-wide analysis of NBS-LRR-encoding genes in., 2003, 15(4): 809-834.
[14] HEATH M C. Hypersensitive response-related death., 2000, 44(3): 321-334.
[15] MALECK K, LEVINE A, EULGEM T, MORGAN A, SCHMID J, LAWTON K A, DANGL J L, DIETRICH R A. The transcriptome ofduring systemic acquired resistance., 2000, 26(4): 403-410.
[16] CARTIEAUX F, THIBAUD M C, ZIMMERLI L, LESSARD P, SARROBENT C, DAVID P, GERBAUD A, ROBAGLIA C, SOMERVILLE S, NUSSAUME L. Transcriptome analysis ofcolonized by a plant-growth promoting rhizobacterium reveals a general effect on disease resistance., 2003, 36(2): 177-188.
[17] CORTES-BARCO A M, HSIANG T, GOODWIN P H. Induced systemic resistance against three foliar diseases ofby (2R, 3R)-butanediol or an isoparaffin mixture., 2010, 157(2): 179-189.
[18] HAAS B J, PAPANICOLAOU A, YASSOUR M, GRABHERR M, BLOOD P D, BOWDEN J, COUGER M B, ECCLES D, LI B, LIEBER M,MacManes M D, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey C N, Henschel R, LeDuc R D, Friedman N, Regev A.transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis.s, 2013, 8(8): 1494-1512.
[19] STOREY J D. The positive false discovery rate: a Bayesian interpretation and the-value., 2003, 31(6): 2013-2035.
[20] SANSEVERINO W, ROMA G, DE SIMONE M, FAINO L, MELITO S, STUPKA E, FRUSCIANTE L, ERCOLANO M R. PRGdb: a bioinformatics platform for plant resistance gene analysis., 2010, 38(Database issue): D814-821.
[21] KANEHISA M, ARAKI M, GOTO S, HATTORI M, HIRAKAWA M, ITOH M, KATAYAMA T, KAWASHIMA S, OKUDA S, TOKIMATSU T, YAMANISHI Y. KEGG for linking genomes to life and the environment.. 2008, 36(Database issue): D480-484.
[22] ZHANG H, ZHAO T, ZHUANG P, SONG Z, DU H, TANG Z, GAO Z. NbCZF1, a novel C2H2-type zinc finger protein, as a new regulator of SsCut-induced plant immunity in., 2016, 57(12): 2472-2484.
[23] GUO Y H, YU Y P, WANG D, WU C A, YANG G D, HUANG J G, ZHENG C C. GhZFP1, a novel CCCH-type zinc finger protein from cotton, enhances salt stress tolerance and fungal disease resistance in transgenic tobacco by interacting with GZIRD21A and GZIPR5.,2009, 183(1): 62-75.
[24] GAO J, CHEN Z, LUO M, PENG H, LIN H J, QIN C, YUAN G S, SHEN Y, DING H P, ZHAO M J, PAN G T, ZHANG Z M. Genome expression profile analysis of the maize sheath in response to inoculation to., 2014, 41(4): 2471-2483.
[25] VENU R C, JIA Y L, GOWDA M, JIA M H, JANTASURIYARAT C, STAHLBERG E, LI H M, RHINEHEART A, BODDHIREDDY R, SINGH P, RUTGER N, KUDRNA D, WING R, NELSON J C, WANG G L. RL-SAGE and microarray analysis of the rice transcriptome afterinfection., 2007, 278(4): 421-431.
[26] WANG Y, KWON S J, WU J N, CHOI J Y, LEE Y H, AGRAWAL G K, TAMOGAMI S, RAKWAL R, PARK S R, KIM B G, JUNG K H, KANG K Y, KIM S G, KIM S T. Transcriptome analysis of early responsive genes in rice duringinfection., 2014, 30(4): 343-354.
[27] YAN Y, JIA H H, WANG F, WANG C, LIU S C, GUO X Q. Overexpression ofreduces tolerance to drought stress and resistance toinfection in transgenic.,2015, 6: Article 265.
[28] YANG S, LI J, ZHANG X, ZHANG Q, HUANG J, CHEN J Q, HARTL D L, TIAN D. Rapidly evolvinggenes in diverse grass species confer resistance to rice blast disease., 2013, 110(46): 18572-18577.
[29] ROBATZEK S, CHINCHILLA D, BOLLER T. Ligand-induced endocytosis of the pattern recognition receptor FLS2 in., 2006, 20(5): 537-542.
[30] HONG S W, JON J H, KWAK J M, NAM H G. Identification of a receptor-like protein kinase gene rapidly induced by abscisic acid, dehydration, high salt, and cold treatments in., 1997, 113(4): 1203-1212.
[31] GODIARD L, SAUVIAC L, TORII K U, GRENON O, MANGIN B, GRIMSLEY N H, MARCO Y. Erecta, an lrr receptor-like kinase protein controlling development pleiotropically affects resistance to bacterial wilt., 2003, 36(3): 353-365.
[32] PARROTT D L, HUANG L, FISCHER A M. Downregulation of a barley () leucine-rich repeat, non-arginine-aspartate receptor-like protein kinase reduces expression of numerous genes involved in plant pathogen defense., 2016, 100: 130-140.
[33] HALTER T, IMKAMPE J, MAZZOTTA S, WIERZBA M, POSTEL S, BÜCHERL C, KIEFER C, STAHL M, CHINCHILLA D, WANG X F, NURRNBERGER T, ZIPFEL C, CLOUSE S, BORST J W, BOEREN S, DE VRIES S C, TAX F, KEMMERLING B. The leucine-rich repeat receptor kinase bir2 is a negative regulator of bak1 in plant immunity., 2014, 24(2): 134-143.
[34] THEODOULOU F L. Plant ABC transporters., 2000, 1465(1/2): 79-103.
[35] 张婧, 陈梦词, 马清, 未丽, 王锁民. 植物ABCG转运蛋白研究进展. 草业学报, 2015, 24(7): 180-188.
ZHANG J, CHEN M C, MA Q, WEI L, WANG S M. Review of advances in the study of plant ABCG transporters., 2015, 24(7): 180-188. (in Chinese)
[36] LIU J, LI W, NING Y, SHIRSEKAR G, CAI Y, WANG X, DAI L, WANG Z, LIU W, WANG G L. The U-box E3 ligase SPL11/PUB13 is a convergence point of defense and flowering signaling in plants., 2012, 160(1): 28-37.
[37] GONZÁLEZ-LAMOTHE R, TSITSIGIANNIS D I, LUDWIG A A, PANICOT M, SHIRASU K, JONES J D. The U-box protein CMPG1 is required for efficient activation of defense mechanisms triggered by multiple resistance genes in tobacco and tomato., 2006, 18(4): 1067-1083.
[38] JIAO L, ZHANG Y L, LU J. Overexpression of a stress-responsive U-box protein geneaffects the accumulation of resistance related proteins in‘Thompson Seedless’.,2017, 112: 53-63.
[39] GUO M, LIU J H, MA X, LUO D X, GONG Z H, LU M H. The plant heat stress transcription factors (hsfs): structure, regulation, and function in response to abiotic stresses., 2016, 7(273): Article 114.
[40] Kornas A, Kuzniak E, Slesak I, Miszalski Z. The key role of the redox status in regulation of metabolism in photosynthesizing organisms., 2010, 57(2): 143-151.
[41] REZAEI M K, SHOBBAR Z S, SHAHBAZI M, ABEDINI R, ZARE S. Glutathione S-transferase (GST) family in barley: identification of members, enzyme activity, and gene expression pattern., 2013, 170(14): 1277-1284.
[42] Liebrand T W, van den Berg G C, Zhang Z, Smit P, Cordewener J H, America A H, Sklenar J, Jones A M, Tameling W I, Robatzek S, Thomma B P, JOOSTEN M H. Receptor-like kinase SOBIR1/EVR interacts with receptor-like proteins in plant immunity against fungal infection., 2013, 110(24): 10010-10015.
[43] CHEONG Y H, KIM M C. Functions of MAPK cascade pathways in plant defense signaling., 2010, 26(2): 101-109.
[44] PEÑA-CORTÉS H, BARRIOS P, DORTA F, POLANCO V, SÁNCHEZ C, SÁNCHEZ E, RAMÍREZ I. Involvement of jasmonic acid and derivatives in plant response to pathogen and insects and in fruit ripening., 2005, 23(3): 246-260.
[45] BELLINCAMPI D, CERVONE F, LIONETTI V. Plant cell wall dynamics and wall-related susceptibility in plant-pathogen interactions., 2014, 5: Article 228.
[46] MATIĆ S, BAGNARESI P, BISELLI C, ORRU’ L, CARNEIRO G A, SICILIANO I, VALÉ G, GULLINO M L, SPADARO D. Comparative transcriptome profiling of resistant and susceptible rice genotypes in response to the seedborne pathogen., 2016, 17(1): 608.
[47] Orshinsky A M, HU J, OPIYO S O, Reddyvari-Channarayappa V, MITCHELL T K, BOEHM M J. Rna-seq analysis of the,–creeping bentgrass pathosystem., 2012, 7(8): e41150.
(责任编辑 岳梅)
Transcriptome analysis of creeping bentgrass ()infected with
SHI Yi, NIU KuiJu, MA Huiling
(College of Pratacultural Science, Gansu Agricultural University/Key Laboratory of Grassland Ecosystem, Ministry of Education/Sino-U.S. Center for Grazingland Ecosystem Sustainability, Lanzhou 730070)
【Objective】The objective of this study is to reveal the gene expression pattern of creeping bentgrass () after been inoculated with, by comparing the gene expression level between plant with disease and without disease during gene transcription, and to identify the key genes of turfgrass responding to pathogen infection. 【Method】which was grown for 14 days,was inoculated withLeaf samples of plant with or without disease were collected after 3 days. High-throughput sequencing technology and Trinity software were used for obtaining the transcriptome. Different expression genes were screened with |log2(fold change)|>1,-value<0.005 as threshold and the assembly was used as reference. Bioinformatics software was used to analyze the transcriptome difference. iTAK was used for transcription factors. All the DEGs were blasted with plant resistance gene database to find the R protein. Mapman software was used to analyze the signaling pathway. 【Result】Using the high-throughput transcriptome sequencing,125 253 092 were obtained. 466 761 transcripts were assembled by Trinity software. More than half of them were longer than 700 bp and N50=1 100 bp. CD-HIT was used to choose 334 212 transcripts as Unigene with the average length of 573 bp and N50=791 bp. Bycomparing thetranscripts with disease and without disease, 7 937up-regulated genes and 1 570 down-regulated genes were obtained. Among the up-regulated genes, 296 of them are transcription factors (TFs), while 142 of down-regulated genes can be defined as TFs. Those TFs were classified as 58 families, including 54 zinc-finger protein containing TFs C2H2, which was the most, then 22 C3H TFs. A total of 451 of different expression genes (DEGs) can be annotated as plant R-protein, which can be classified into 33 families. Among these families, NBS-LRR containing resistance protein, LRR-like receptor protein kinase, ABC-2 type transporter, U-box domain containing protein kinase, and heat shock protein gene, these 5 families showed the most variation. A bunch of up-regulated DEGs can be mapped in plant biotic response pathway and enriched in pathogen recognition, ROS eliminate, signaling transport, programmed cell death, pathogenesis-related protein and so on, while the down-regulated genes were enriched in plant growth and development pathways. The expression level of random selected DEGs was tested with qRT-PCR, and they were consistent with the result of RNA-seq analysis. The random selected genes included 12 C2H2 TFs, 10 C3H TFs and 12 R-protein genes.【Conclusion】The inoculation of pathogen caused great expression difference oftranscriptome. Most of the TFs, R-protein and defense-related genes were up-regulated to suppress pathogen growth. The inherent resistance oftohas been generated with the co-action of all these genes.
;; transcriptome; different expression genes; disease resistance mechanism
2017-03-06;接受日期:2017-04-27
国家自然科学基金(31360583)
史毅,E-mail:shiyi214@126.com。通信作者马晖玲,E-mail:mahl@gsau.edu.cn
猜你喜欢
浙江农林大学学报(2022年5期)2022-10-12
湖北农业科学(2022年11期)2022-07-18
实用肿瘤学杂志(2020年4期)2020-12-08
心电与循环(2020年1期)2020-02-27
陕西农业科学(2019年2期)2019-04-12
中国酿造(2017年11期)2017-12-06
江苏农业科学(2017年5期)2017-04-15
植物保护(2015年1期)2015-02-14
中国老年学杂志(2015年9期)2015-01-31
湖北农业科学(2014年3期)2014-07-21
