
甜菜WRKY转录因子全基因组鉴定及其在非生物胁迫下的表达分析
2017-09-26 08:02孔维龙于坤但乃震杨绍宗包满珠黄向荣傅小鹏
中国农业科学 2017年17期
孔维龙,于坤,但乃震,杨绍宗,包满珠,黄向荣,傅小鹏
甜菜WRKY转录因子全基因组鉴定及其在非生物胁迫下的表达分析
孔维龙1,于坤2,但乃震1,杨绍宗1,包满珠1,黄向荣1,傅小鹏1
(1华中农业大学园艺林学学院/园艺植物生物学教育部重点实验室,武汉 430070;2石河子大学农学院/新疆生产建设兵团绿洲生态农业重点实验室,新疆石河子832003)
【目的】WRKY转录因子是一类植物响应生物、非生物胁迫,对生长发育都起重要调控作用的转录因子。在甜菜全基因组信息分析的基础上,鉴定WRKY家族基因(),解析其组织特异性及盐、热胁迫下的表达情况,为该类基因的功能研究提供参考,为观赏甜菜和石竹目其他观赏植物的基因工程打下基础。【方法】以75条拟南芥WRKY蛋白为参考,根据WRKY保守蛋白序列(PF03106)利用hmm和BLAST同源性搜索对甜菜WRKY家族基因进行鉴定。利用MapInspect、GSDS2.0、MEGA5.0、DNAMAN5.0、WebLogo 3、MEME生物信息学工具对甜菜WRKY家族基因染色体定位、系统发生关系、基因结构、蛋白质保守结构域、保守元件进行预测和分析。利用RNA-seq和qRT-PCR分析甜菜组织表达特异性,盐胁迫、热胁迫条件下表达情况。【结果】甜菜WRKY家族基因包含40个成员,其中39条不均匀地分布在9条染色体上,另外1条定位到随机片段上。根据WRKY保守域特征并与拟南芥WRKY蛋白进化分析,可将40个成员分为Ⅰ、Ⅱ、Ⅲ 3类,Ⅰ类有9个成员,Ⅱ类有26个成员,Ⅲ类有5个成员。根据进化关系Ⅱ类可进一步分为Ⅱa(1个)、Ⅱb(4个)、Ⅱc(9个)、Ⅱd(5个)和Ⅱe(7个)5个亚类。基因结构分析发现,甜菜外显子和内含子数目具有高变异性(2—7个外显子),即使同一亚类内也都差异较大。保守元件分析显示同一类或亚类内成员具有相同的保守元件。WRKY保守域分析发现2个WRKY七肽域变型:WRKYGKK和WRKYGEK。每个至少在2个组织中表达,30个在叶中表达,40个在花序中均有表达,36个在幼叶中有表达,38个在直根中有表达,39个在幼苗中有表达,36个在种子中有表达。各表达量差异较大,可分为低表达、高表达基因两类,如、、、、、、、、、和在各组织中均有较高表达,而、、、、和在各组织中均表达较低。热胁迫条件下、、、、、和上调表达;盐胁迫条件下、、、和呈现不同程度上调表达;对热、盐2种胁迫均有明显响应。【结论】甜菜WRKY蛋白结构高度保守,基因序列长度和内含子数量变化很大,在不同组织中呈现出多种表达模式,部分响应热或盐胁迫,对甜菜逆境生理调控起重要作用。
甜菜;WRKY转录因子;生物信息学;胁迫;表达分析
0 引言
【研究意义】甜菜()是温带地区重要的产糖作物,据联合国粮食农业组织(FAO)统计,世界每年30%的植物糖产量均出自于甜菜,同时它也是生产生物乙醇、天然色素、食用蔬菜及动物饲料的重要来源,近年来越来越多的甜菜栽培种(红梗叶甜菜、红叶甜菜)作为观赏植物被广泛应用于景观配置[1]。WRKY作为一类对植物生长发育、逆境响应都有重要作用的转录因子,对其进行全面的生物信息学分析和表达特异性研究,对甜菜栽培、育种、生产推广以及功能研究具有重要意义。【前人研究进展】WRKY家族是最大的转录因子之一,因高度保守的WRKY保守域而得名。WRKY保守域能够特异识别并结合靶基因启动子区域的W盒[C/T]TGAC[T/C]和SURE元件(糖响应顺式元件)。WRKY保守域包含大约60个氨基酸,由1个N端WRKYGQK七肽域和C端锌指结构构成[2]。WRKYGQK七肽域又有WRKYGKK、WRKYDQK和WRKYDHK[3]等多种变型;锌指结构有C2H2(CX4-5CX22-23HX1H)和C2HC(CX7CX23HX1C)2个类型。除WRKYGQK七肽域和锌指结构外,一些WRKY蛋白还含有亮氨酸拉链结构(LZ)、核定位信号域(NLS)、丝氨酸/苏氨酸富集域、谷氨酰胺富集域和脯氨酸富集域[4-5]。根据WRKY域数量和锌指结构类型,WRKY家族可被分为Ⅰ、Ⅱ、Ⅲ 3类。第Ⅰ类包含2个WRKY七肽域和1个C2H2型锌指结构;第Ⅱ类包含1个WRKY七肽域和1个C2H2类型锌指结构;第Ⅲ类包含1个WRKY七肽域和1个C2HC型锌指结构。第Ⅱ类根据进化关系可细分成Ⅱa+b、Ⅱc和Ⅱd+e 3类[6]。最先报导的WRKY转录因子是甘薯的[7],随后相继在马铃薯[8]、烟草[9]、小麦和大麦[10-11]、拟南芥[12-16]、胡椒[17]、水稻[18-20]、辣椒[21]、杨树[22]、油菜[23]、黄瓜[24]、棉花[25]、胡萝卜[26]、番木瓜[27]、柳树[28]、苹果[29]、谷子[30]等鉴定得到WRKY转录因子。大量研究表明,WRKY转录因子作为正调蛋白或负调蛋白对植物应答生物与非生物胁迫起调节作用。除此之外,也参与调控植物生理发育过程,涉及激素信号转导、生物合成调节、叶片衰老、胚形成及种子萌发等多个方面[5,31]。如拟南芥的WRKY转录因子应答脱落酸和干旱胁迫[32]。超量表达和的转基因小麦在耐盐性、耐旱性和抗冻性等方面均有提高[33]。在水稻糊粉细胞,WRKY转录因子还能调控GAMYB(GA转录因子激活子)介导GA信号途径[34]。【本研究切入点】甜菜是重要的产糖作物,同时观赏甜菜和石竹目其他观赏植物,如香石竹()、石竹()、须苞石竹()等被广泛应用于室内装饰、园林造景等领域。但它们生长发育均容易受多种生物、非生物逆境的影响,严重影响甜菜产量,限制观赏甜菜和石竹目其他观赏植物的应用和推广。而目前石竹目植物高质量的基因组和转录组可利用数据相对有限,仅甜菜具有高质量的基因组和转录组数据可供利用。【拟解决的关键问题】以甜菜全基因组为基础,鉴定WRKY家族成员,分析基因结构、染色体定位、亚细胞定位、蛋白保守结构域及系统发生关系等信息,明确甜菜WRKY家族基因在各个组织中的表达特征和多种胁迫条件下的表达模式。为WRKY转录因子功能的解析和抗逆调控网络的构建打下基础,同时预测出与观赏甜菜抗逆性紧密相关的候选,为观赏甜菜以及石竹科其他观赏植物抗逆分子育种提供依据。
1 材料与方法
1.1 材料
试验于2016年12月至2017年2月在华中农业大学园艺植物生物学教育部重点实验室完成,供试品种为观赏甜菜(var.),购于北京东升种业有限公司。播种于人工气候箱(达斯卡特RGL-P,合肥),光照强度16 000 lx,光周期为16 h光照/8 h黑暗,培养温度22℃,相对湿度65%。待小苗长至6—7片叶时,进行盐和高温非生物胁迫处理。用300 mmol·L-1NaCl溶液浇灌植株根部以模拟盐胁迫,设置清水浇灌为对照,2日后取顶部第2、3片幼叶样品迅速放入液氮保存。高温处理方式:38℃处理2 h,之后取顶部第2、3片幼叶迅速放入液氮保存。所有胁迫处理均于人工气候箱进行,对照和处理分设3次重复,每个重复6株。
1.2 甜菜WRKY家族基因的鉴定
使用hmm和BLAST同源性搜索两种方法来鉴定WRKY转录因子,WRKY蛋白结构域信息(PF03106)来自Pfam数据库(http://pfam.xfam.org/),利用该结构域HMM文件,用HMMER 3.0软件在甜菜功能基因组数据库(http://www.genomforschung.uni-bielefeld. e/en/projects/annobeet)中进行的对比搜索[29],参数设置为默认参数。下载已鉴定的75条拟南芥WRKY蛋白序列,利用在线基因组BlastP搜索,再次搜索WRKY转录因子。去除重复序列,利用Pfam和SMART(http://smart.embl-heidelberg.de/)对候选基因进行进一步确认,最终获得甜菜WRKY家族基因。
1.3 甜菜WRKY家族基因的生物信息分析
甜菜WRKY家族基因内含子、外显子和基因组定位信息均来自于甜菜基因组数据库,利用在线软件GSDS2.0(http://gsds.cbi.pku.edu.cn/index.php)绘制基因结构图,运用MapInspect工具绘制染色体定位图;利用序列分析软件DNAMAN 5.0和在线软件WebLogo 3(http://weblogo.threeplusone.com/)分析甜菜WRKY蛋白的WRKY保守域;利用在线软件 MEME(http://meme-suite.org/tools/meme)对甜菜WRKY蛋白进行基序预测,参数中预测数目设置为8,其他参数均为默认设置;利用MEGA5.0软件对甜菜WRKY家族蛋白序列与拟南芥WRKY蛋白序列(每个家族/亚家族随机选取2个)进行比对,用邻位(Neighbor-Joining)算法构建系统发生树,校验参数Bootstrap重复1 000次。
1.4 甜菜表达分析
RNA-seq数据来自于SRA(SRX287608-287615;SRX647324;SRX647712;SRX647714)(https://www. cbi.nlm.nih.gov/sra/)[35],分析甜菜在不同组织(幼苗、直根、幼叶、叶、花序、种子)中的表达情况以及各在盐胁迫、热胁迫下幼叶中表达变化。具体方法:用HISAT2将reads比对到参考基因组上,用StringTie计算基因表达量[36]。
RNA-seq测序用KWS2320植株生长于沙滩,生长温度21℃,光周期为10 h光照/14 h黑暗,光照强度4 000—6 000 lx,相对湿度60%。测序样品为花序、直根、基部叶片、幼叶(顶端第3、4片幼叶不含叶中脉)、种子样品,幼苗为20℃培养2 d的出芽苗[1,36]。盐胁迫处理:植株全营养液水培,23 d苗龄时转入含50 mmol·L-1NaCl的全营养液,每天增加50 mmol·L-1NaCl浓度,逐渐增加NaCl浓度到300 mmol·L-1,此后保持300 mmol·L-1NaCl全营养液胁迫2 d,于第30天采取幼叶样品。热胁迫处理:植株全营养液水培30 d,幼叶样品采摘前进行35℃高温胁迫3 h。对照幼叶取材于全营养水培30 d的植株。所有样品使用Nucleospin RNA Plant kit试剂盒(Macherey-Nagel,德国)进行提取,利用Hiseq 1500测序仪进行双末端测序[35-36]。
采用EASYspin植物RNA快速提取试剂盒(艾德莱生物科技有限公司,北京)和TRUEscript 1st Strand cDNA Synthesis Kit With gDNA Eraser反转录试剂盒(艾德莱生物科技有限公司,北京)提取样品RNA反转录成cDNA。使用Primer 5软件设计实时定量PCR(qRT-PCR)特异性引物,由上海生工生物工程有限公司合成(表1)。

表1 荧光定量PCR扩增引物序列
使用ABI公司(美国)Q7实时定量仪进行荧光定量分析,定量试剂选用宝生物工程(大连)有限公司SYBR Premix EX TaqTMII,以作为内参基因(KF214784)[37]。反应体系(10 μL)为SYBR染料5 μL、DyII 0.2 μL、ddH2O 3 μL、上下游引物各0.4 μL、cDNA 1 μL。反应条件为95℃ 4 min;95℃ 20 s,60℃20 s,72℃ 40 s,40个循环,每个处理3次重复。采用2-ΔΔCT法进行数据分析,基因的相对表达量使用平均值(mean)±标准误(SE)表示。
2 结果
2.1 甜菜WRKY家族基因的鉴定与系统发育分析
共筛选得到40个甜菜(表2),根据Eulgem等[2]和Rushton等[31]描述的WRKY保守域,可将其分为Ⅰ、Ⅱ、Ⅲ 3类。其中Ⅰ有9个成员;Ⅱ类有26个成员;Ⅲ类有5个成员。参照拟南芥中的分类标准,根据进化关系进一步将Ⅱ类归为Ⅱa(1个)、Ⅱb(4个)、Ⅱc(9个)、Ⅱd(5个)和Ⅱe(7个)亚类。甜菜WRKY蛋白序列与拟南芥(每类或亚类、随机选取两条)进行聚类和系统发生分析的结果(图1)与根据WRKY结构域和锌指类型进行鉴定的结果(表2)一致,Ⅲ类是独立的分支,Ⅰ和Ⅱ类显示相对较近的关系;Ⅱ-a和Ⅱ-b亚类聚类成一个分支,Ⅱ-e和Ⅱ-d亚类聚类成一个分支,Ⅱ-c与I聚类为一个分支。此外,与拟南芥和水稻类似,甜菜Ⅱ类也可分成Ⅱa+b、Ⅱc和Ⅱd+e 3类,整个WRKY被分成5类(图1)。

表2 甜菜WRKY家族基因信息
*为WRKY结构变型* modified WRKY heptapeptide
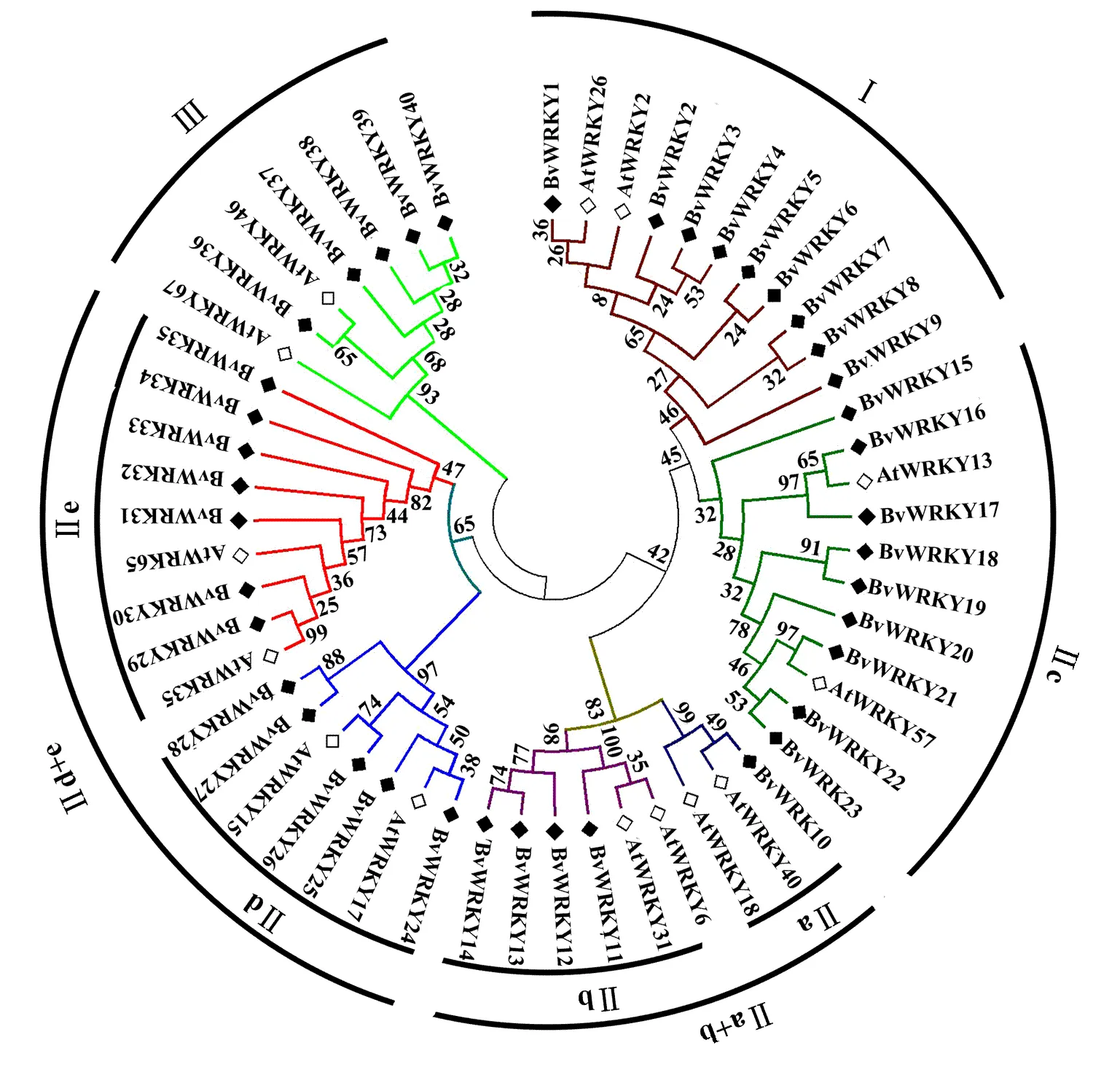
黑色方框代表甜菜WRKY蛋白,白色方框代表拟南芥WRKY蛋白。Ⅰ代表Ⅰ类,Ⅱ-a+b代表Ⅱ-a+b类,Ⅱ-c代表Ⅱ-c类,Ⅱ-d+e代表Ⅱ-d+e类,Ⅲ代表Ⅲ类,Ⅱa代表II a亚类,Ⅱb代表Ⅱb亚类,Ⅱc代表Ⅱc亚类,Ⅱd代表Ⅱd亚类,Ⅱe代表Ⅱe亚类。下同
2.2 甜菜WRKY蛋白WRKY保守域序列分析
对甜菜WRKY保守域进行分析,发现此域高度保守(表2、图2),偶有变型。其中Ⅰ类的C端WRKY七肽域和锌指结构为WRKYGQK和CX4CX23HXH,N端WRKY七肽域和锌指结构为WRKYGQK和CX4CX22HXH(例外,其锌指结构是CX4CX23HXH形式与C段锌指更类似);Ⅱ类a、b、d、e亚类WRKY七肽域和锌指结构为WRKYGQK和CX5CX23HXH(例外,其WRKY七肽域是WRKYGEK形式,为WRKY七肽域变形),Ⅱ类c亚类七肽域和锌指结构为WRKYGQK和CX4CX23HXH(例外,其WRKY七肽域WRKYGKK,为WRKY七肽域变异);Ⅲ类七肽域和锌指结构为WRKYGQK和CX7CX23HXC(图2)。
2.3 甜菜内含子和外显子结构分析
甜菜外显子和内含子数目具有高变异性(2—7个外显子)(图3)。4个含有2个外显子,占10%;23个含有3个外显子,占57.5%;7个含有4个外显子,占17.5%;4个含有5个外显子,占10%;仅有2个含有6个外显子。Ⅲ类和Ⅱd+e类多为3个外显子(和例外,只有2个);Ⅱc、Ⅱa+b和Ⅰ类数量变异性较大,其中Ⅰ类和Ⅱa+b类变异性极大(3—6个外显子)。Ⅲ类和Ⅱd+e类相对保守,而Ⅱc、Ⅱa+b和Ⅰ类内含子外显子结构分化严重,变化较大。

Ⅰ类被分为Ⅰ-C亚类和Ⅰ-N亚类,Ⅱ-a, b代表Ⅱa与Ⅱb亚类,Ⅱ-c代表Ⅱ-c亚类,Ⅱ-d代表Ⅱ-d亚类,Ⅱ-e代表Ⅱ-e亚类,Ⅲ代表Ⅲ类
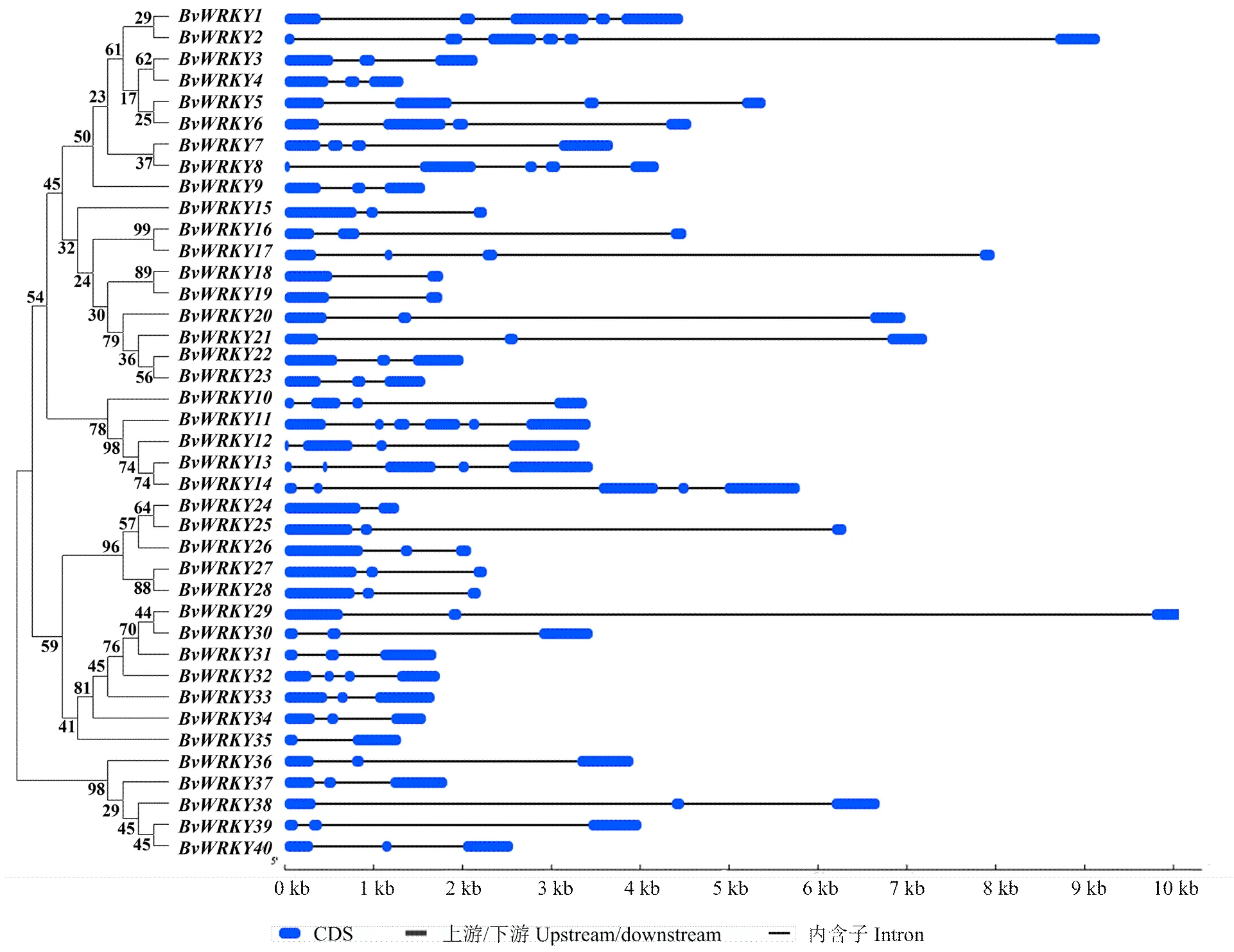
图3 甜菜WRKY内含子和外显子结构
2.4 甜菜染色体定位
染色体定位结果显示,甜菜40个中有39个分布于9条染色体中,且呈不均匀分布,1个()未能定位到任何染色体上(图4),定位在随机片段上。其中第2和6染色体上的分布最多,各有7个,各占17.5%;第5和1染色体次之,各有6、5个,各占15%、12.5%;第9染色体分布最少,仅有2个,占5%;第3、4、7和8染色体均分布3个,各占7.5%(图4)。甜菜各类基因在染色体上的分布也存在差异,其中Ⅱd+e类分布最为广泛,除第7染色体之外,其他8条染色体上均有分布;Ⅰ类分布于第2、4、6、7、9染色体上;Ⅱc类分布于第1、2、3、5、6染色体上,并有一条未能定位到任何染色体上;Ⅲ类分布于第1、2、3、5、7染色体上;Ⅱa+b类分布于第2、5、6、8染色体上(图4、表2)。甜菜在9条染色体上的分布较多,进一步对其进行串联重复情况分析。以2个或多个同源基因(属于相同的家族基因或类)在染色体上的物理位置不超过100 kb,彼此之间相似度大于40%,即为串联重复基因为标准[29,38-39],并没有发现任何串联重复和片段重复情况,即使有些基因彼此之间物理距离较近,如和物理距离为小于100 kb,也因不属于同一类,排除其串联重复的可能。
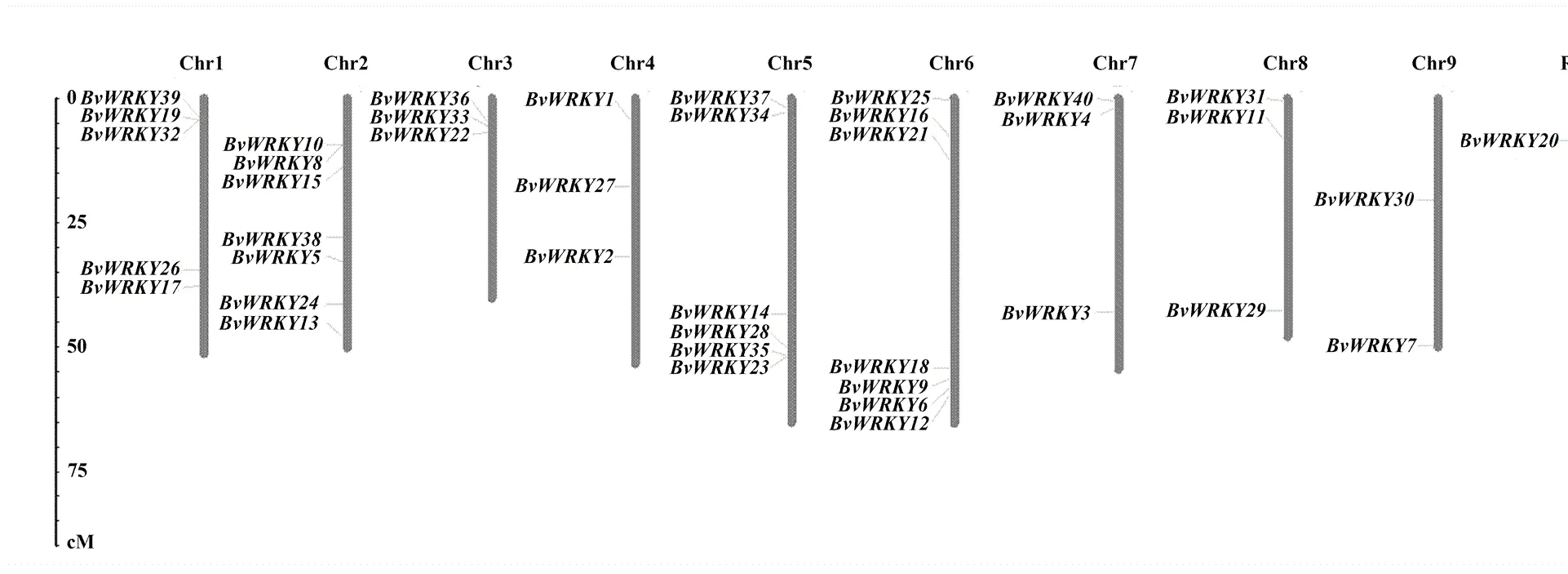
图4 甜菜WRKY染色体定位
2.5 甜菜WRKY家族基因保守元件
分析保守元件预测结果,Motif 1和Motif 3包含WRKY七肽域,Motif 2和Motif 4为锌指结构域,Motif 1和Motif 2构成C端WRKY盒;Motif 3和Motif 4构成N端WRKY盒。Motif 6是亮氨酸拉链盒(BRLZ domain),Motif 7是苏氨酸富集域,Motif 5和Motif 8为未知盒。Ⅰ类具有2个WRKY盒,具有Motif 1—Motif 4共4个motif,而其他类只具有C端WRKY盒,即仅具有Motif 1和Motif 2,无Motif 3和Motif 4。Motif 5几乎存在于Ⅰ类、Ⅱc类的每一个成员中(除BvWRKY 7外),部分存在于Ⅱd+e、Ⅲ中,而Ⅱa+b类中无Motif 5;Motif 6和Motif 8特异地存在于Ⅱa+b类中;Motif 7仅偶然存在于Ⅱa+b、II c、Ⅱd+e中(图5)。各类motif存在数量和种类较为固定,与系统发育树结果(图1)和鉴定分类结果(表2)一致。
2.6 甜菜组织特异性表达谱
分析根、叶、幼叶、花序、幼苗和种子6个组织的RNA-seq数据,发现每个至少在2个组织中表达。其中,30个在叶中表达,40个在花序中均有表达,36个在幼叶中有表达,38个在直根中有表达,39个在幼苗中有表达,36个在种子中有表达。40个在各组织中的表达量存在较大差异,直根中总表达量最高(RPKM=371.05);在幼叶、种子、幼苗中次之,RPKM分别为311.47、251.17、208.00;花序、叶中表达最少,RPKM分别为142.07、119.33。6个组织的表达谱聚类发现,花序、幼叶和叶中的表达较为类似;种子、幼苗和直根中的表达较为类似(图6)。
通过聚类分析,40个基因在各组织中的表达结果可明显的分为两组,一组(A组)包括22个成员,为高表达基因;另一组(B组)包括18个成员,为低表达基因。A组中所有基因在各组织中均有表达,但各成员之间表达模式差异较大,一部分基因在各个组织中均有较高表达量,如、、、、、、、、、和。一部分基因呈现组织表达特异性,如优势表达于幼苗和种子;优势表达于直根和幼苗;在叶中优势表达。B组中有6个基因在各个组织中均表达量较低,分别为、、、、和。12个基因在某些组织中优势表达,其他组织中表达量极低。如、和优势表达于直根;优势表达于幼叶;优势表达于种子(图6)。
2.7 甜菜在2种非生物胁迫下的表达谱
对热胁迫和盐胁迫处理后幼叶RNA-seq结果进行分析。热胁迫之后,大部分下调表达,其中、下调最为明显;有7个呈现不同程度的上调表达,其中、、、和上调较为明显(图7)。盐胁迫条件下,绝大多数出现不同程度的下调表达,其中下调最为明显。少数基因呈现上调表达,其中和上调表达最为明显。此外,在热胁迫和盐胁迫后均出现上调表达,推测对热和盐2种非生物逆境均有积极响应作用(图7)。
2.8 甜菜的qRT-PCR表达分析
根据RNA-seq分析结果,盐胁迫下5个基因表达量明显上调,热胁迫下7个基因上调表达,这些基因被初步认定为抗逆相关候选基因(表1)。利用qRT-PCR进一步分析6—7片叶龄的观赏甜菜中这些候选基因在盐胁迫和热胁迫下的表达模式。结果表明,盐胁迫条件下、、、、3表达量上调;热胁迫下、、、、、、表达量上调(图8),与RNA-seq结果基本一致。略有不同的是盐胁迫和热胁迫下,qRT-PCR分析结果中基因上调程度均没有RNA-seq分析结果高。究其原因,可能与甜菜品种、胁迫方式及胁迫强度有关。
3 讨论
甜菜属于真双子叶植物的基部类群,对其WRKY转录因子系统的鉴定填补了被子植物到核心双子叶植物WRKY转录因子无人报道的空缺。本研究共鉴定出40个甜菜WRKY转录因子,不存在缺失类和亚类等情况。其的数量与被子植物基部植物的挪威云杉(32个)[26]和核心双子叶植物加拿大油菜(43个)[23]以及蓖麻子(47个)[40]相接近。相较其他双子叶植物以及单子叶植物差别较大,如杨树有104个[41]、大豆有188个[42]、棉花有116个[43]、大白菜有145个[44]、苹果有139个[26]、拟南芥有74个[5],均远多于甜菜。究其原因,发现这些物种都经历过基因重复事件,包括全基因组复制、串联复制和片段复制。如白菜在进化过程中经历六倍体阶段,由全基因组复制事件产生3个亚基因组,导致白菜大量基因发生扩张。另外,花生(75个)[45]、苹果[29]、水稻[26]、拟南芥[5]、马铃薯(81个)[26]、番茄(81个)[46]均发生过串联复制和片段复制,导致基因数量增多。相比于上述物种,甜菜既没有发生过全基因组的复制事件,WRKY转录因子又无串联或片段复制发生,这是其家族基因数量较少的重要原因。
WRKY转录因子WRKYGOK基序与DNA的“大沟”结合,进而与W-box的T上的甲基原子团进行非极性结合识别DNA。WRKYGOK基序的变异将导致与T-box结合能力大幅下降甚至完全丧失。在甜菜中发现2个WRKY七肽域变型:的WRKYGKK七肽域和的WRKYGEK七肽域。在番茄[38]、桃树[47]中亦有WRKYGKK七肽域,而WRKYGEK七肽域却少有报道。大豆中含有WRKYGKK变体的和不能正常结合W-box[48]。具有WRKYGKK变体的烟草的识别另一结合序列TTTTCCAC而不是正常Wbox[49]。推测甜菜2个WRKY七肽域变型可能使WRKY蛋白失去与DNA的识别和结合能力,或者能识别其他新基序,产生新的功能。
分析组织表达特征,发现甜菜在各组织表达差异较大,总表达量在根部最高,在叶表达量最低,大多数基因在根中优势表达,如、和。类似的,丹参绝大多数(22/61)也在根中呈现优势表达,总表达量较其他组织高[50]。水稻也在根优势表达,试验证明磷酸盐匮乏的处理条件下,超量表达的水稻Pi含量较对照高6%,分蘖数、谷粒重量均高于对照,证明能提高水稻对磷酸盐的吸收[51]。拟南芥、亦在根中优势表达,其超量表达植株亦被证明能增加根Pi含量和提高Pi吸收率[52-53]。推测甜菜在根中优势表达部分也可能涉及磷酸盐吸收与转运途径。
RNA-seq和qRT-PCR的结果均表明、、、、表达量受盐胁迫诱导而上调表达;、、、、、表达量受热胁迫诱导而上调表达。在水稻、小麦、葫芦卜、马铃薯等物种中亦有转录因子响应盐、热、冷、干旱胁迫上调表达的报道[26,54-56],观赏甜菜的在盐和热胁迫条件下均被诱导上调表达,表明转录因子家族对植物适应胁迫环境有重要作用,与植物逆境生长密切相关,部分WRKY转录因子响应多种胁迫。
4 结论
甜菜WRKY家族基因序列长度和内含子数量变化很大,但蛋白结构高度保守;在不同组织中呈现出多种表达模式。RNA-seq和qRT-PCR分析发现、、、和受盐胁迫诱导表达上调,、、、、、和受热胁迫诱导表达上调,推测这些基因可能参与了甜菜非生物逆境下生长发育的调控。
References
[1] Dohm J C, Minoche A E, Holtgräwe D, Capellagutiérrez S, Zakrzewski F, Tafer H, Rupp O, Sörensen T R, Stracke R, Reinhardt R. The genome of the recently domesticated crop plant sugar beet ()., 2014, 505(7484): 546-549.
[2] Eulgem T, Rushton P J, Robatzek S, Somssich I E. Thesuperfamily of plant transcription factors., 2000, 5(5): 199-206.
[3] Guo C, Guo R, Xu X, Gao M, Li X, Song J, Zheng Y, Wang X. Evolution and expression analysis of the grape (L.)gene family., 2014, 65(6): 1513-1528.
[4] Cormack R S, Eulgem T, Rushton P J, Köchner P, Hahlbrock K, Somssich I E. Leucine zipper-containing WRKY proteins widen the spectrum of immediate early elicitor- inducedtranscription factors in parsley., 2002, 1576(1/2): 92-100.
[5] Somssich I E.transcription factors: from DNA binding towards biological function., 2004, 7(5): 491-498.
[6] Zhang Y, Wang L. Thetranscription factor superfamily: its origin in eukaryotes and expansion in plants., 2005, 5(1): 1-12.
[7] Ishiguro S, Nakamura K. Characterization of a cDNA encoding a novel DNA-binding protein,, that recognizessequences in the 5' upstream regions of genes coding for sporamin and beta-amylase from sweet potato., 1994, 244(6): 563-571.
[8] Dellagi A, Birch P J, Heilbronn J, Avrova A O, Montesano M, Palva E T, Lyon G D. A potato gene,, is rapidly induced byssp.,, ethylene and salicylic acid., 2000, 157(2): 201-205.
[9] Yoda H, Ogawa M, Yamaguchi Y, Koizumi N, Kusano T, Sano H. Identification of early-responsive genes associated with the hypersensitive response to tobaccoand characterization of a-type transcription factor in tobacco plants., 2002, 267(2): 154-161.
[10] Sun C, Palmqvist S, Olsson H, Borén M, Ahlandsberg S, Jansson C. A noveltranscription factor,, participates in sugar signaling in barley by binding to the sugar- responsive elements of the iso1 promoter., 2003, 15(9): 2076-2092.
[11] 刘自成, 苗丽丽, 王景一, 杨德龙, 毛新国, 景蕊莲. 普通小麦转录因子基因的克隆及功能分析. 中国农业科学, 2016, 49(12): 2245-2254. Liu Z C, Miao L L, Wang J Y, Yang D L, Mao X G, Jing R L. Cloning and characterization of transcription factor)., 2016, 49(12): 2245-2254. (in Chinese)
[12] Chen H, Lai Z, Shi J, Xiao Y, Chen Z, Xu X. Roles of,andtranscription factors in plant responses to abscisic acid and abiotic stress., 2010, 10(1): 443-462.
[13] Chen W, Provart N J, Glazebrook J, Katagiri F, Chang H S, Eulgem T, Mauch F, Luan S, Zou G, Whitham S A, BUDWORTH P R, YAO T, XIE Z, CHEN X, LAM S, KREPS J A, HARPER J F, SI-AMMOUR A, MAUCH-MANI B, HEINLEIN M, KOBAYASHI K, HOHN T, DANGL J L, WANG X, ZHU T. Expression profile matrix oftranscription factor genes suggests their putative functions in response to environmental stresses., 2002, 14(3): 559-574.
[14] Hwang S H, Yie S W, Hwang D J. Heterologous expression ofgene inactivates the expression of defense related genes and enhances resistance to pathogens., 2011, 181(3): 316-323.
[15] Xu X, Chen C, Fan B, Chen Z. Physical and functional interactions between pathogen-induced,, andtranscription factors., 2006, 18(5): 1310-1326.
[16] ZHANG Z Z, QAMAR S A, Chen Z X, Mengiste T.transcription factor is required for resistance to necrotrophic fungal pathogens., 2006, 48(4): 592-605.
[17] Zheng J, Zou X, Mao Z, Xie B. A novel pepper (L.)gene,, is involved in pathogen stress responses., 2011, 54(5): 329-337.
[18] Ramamoorthy R, Jiang S Y, Kumar N, Venkatesh P N, Ramachandran S. A comprehensive transcriptional profiling of thegene family in rice under various abiotic and phytohormone treatments., 2008, 49(6): 865-879.
[19] Christian R, Shen Q x. Thegene family in rice ()., 2007, 49(6): 827-842.
[20] Zhang J, Peng Y l, GUO Z J. Constitutive expression of pathogen-inducibleenhances disease resistance and affects root growth and auxin response in transgenic rice plants., 2008, 18(4): 508-521.
[21] Oh S K, Baek K H, Park J M, Kamoun S, Choi D.proteinis a negative regulator of pathogen defense., 2008, 177(4): 977-989.
[22] Levée V, Major I, Levasseur C, Tremblay L, Mackay J, Séguin A. Expression profiling and functional analysis ofreveals a regulatory role in defense., 2009, 184(1): 48-70.
[23] Yang B, Jiang Y, Rahman M H, Deyholos M K, Kav N N. Identification and expression analysis oftranscription factor genes in canola (L.) in response to fungal pathogens and hormone treatments., 2009, 9(1): 261-279.
[24] Ling J, Jiang W, Zhang Y, Yu H, Mao Z, Gu X, Huang S, Xie B. Genome-wide analysis ofgene family in., 2011, 12(1): 1-20.
[25] Yao D, Zhang X, Zhao X, Liu C, Wang C, Zhang Z, Zhang C, Wei Q, Wang Q, Yan H. Transcriptome analysis reveals salt-stress-regulated biological processes and key pathways in roots of cotton (L.)., 2011, 98(1): 47-55.
[26] Li M Y, Xu Z S, Tian C, Huang Y, Wang F, Xiong A S. Genomic identification oftranscription factors in carrot () and analysis of evolution and homologous groups for plants., 2016, 6(1): 1-17.
[27] Pan L J, Jiang L. Identification and expression of thetranscription factors ofin response to abiotic and biotic stresses., 2014, 41(3): 1215-1225.
[28] Bi C W, Xu Y Q, Ye Q L, Yin T M, Ye N. Genome-wide identification and characterization ofgene family in., 2016, 4: e2437.
[29] 谷彦冰, 冀志蕊, 迟福梅, 乔壮, 徐成楠, 张俊祥, 董庆龙, 周宗山. 苹果基因家族生物信息学及表达分析. 中国农业科学, 2015, 48(16): 3221-3238. Gu Y B, Ji Z R, Chi F M, Qiao Z, Xu C N, Zhang J X, Dong Q L, Zhou Z S. Bioinformatics and expression analysis of thegene family in apple., 2015, 48(16): 3221-3238. (in Chinese)
[30] 祖倩丽, 尹丽娟, 徐兆师, 陈明, 周永斌, 李连城, 马有志, 闵东红, 张小红. 谷子转录因子的分子特性及功能鉴定. 中国农业科学, 2015, 48(5): 851-860.Zu Q L, Yin L J, Xu Z S, Chen M, Zhou Y B, Li L C, Ma Y Z, Min D H, Zhang X H. Molecular characteristics and functional identification of foxtail millet transcription factor., 2015, 48(5): 851-860. (in Chinese)
[31] Rushton P J, Somssich I E, Ringler P, Shen Q J.transcription factors., 2014, 15(5): 247-258.
[32] Ren X, Chen Z, Liu Y, Zhang H, Zhang M, Liu Q, Hong X, Zhu J K, Gong Z. ABO3, atranscription factor, mediates plant responses to abscisic acid and drought tolerance in., 2010, 63(3): 417-429.
[33] Niu C F, Wei W, Zhou Q Y, Tian A G, Hao Y J, Zhang W K, Biao M A, Lin Q, Zhang Z B, Zhang J S. Wheatgenesandregulate abiotic stress tolerance in transgenicplants., 2012, 35(6): 1156-1170.
[34] Zhen X, Zhang Z L, Zou X, Yang G, Komatsu S, Shen Q J. Interactions of two abscisic-acid inducedgenes in repressing gibberellin signaling in aleurone cells., 2006, 46(2): 231-242.
[35] Minoche A E, Dohm J C, Schneider J, Holtgräwe D, Viehöver P, Montfort M, Sörensen T R, Weisshaar B, Himmelbauer H. Exploiting single-molecule transcript sequencing for eukaryotic gene prediction., 2015, 16(1): 1-13.
[36] Stracke R, Holtgräwe D, Schneider J, Pucker B, Sörensen T R, Weisshaar B. Genome-wide identification and characterization ofgenes in sugar beet ()., 2014, 14(1): 1-17.
[37] 梁娜, 伍国强, 冯瑞军, 刘左军, 李志忠. 甜菜基因片段的克隆及序列分析. 中国糖料, 2014(4): 13-15.Liang N, Wu G J, Feng R J, Liu Z J, Li Z Z. Cloning and sequence analysis ofgene fragments from sugarbeet., 2014(4): 13-15. (in Chinese)
[38] Huang S, Gao Y, Liu J, Peng X, Niu X, Fei Z, Cao S, Liu Y. Genome-wide analysis oftranscription factors in., 2012, 287(6): 495-513.
[39] Hu L, Liu S. Genome-wide analysis of thegene family in cucumber., 2012, 55(3): 245-256.
[40] Li H L, Zhang L B, Guo D, Li C Z, Peng S Q. Identification and expression profiles of thetranscription factor family in., 2012, 503(2): 248-253.
[41] He H, Dong Q, Shao Y, Jiang H, Zhu S, Cheng B, Xiang Y. Genome-wide survey and characterization of thegene family in., 2012, 31(7): 1199-1217.
[42] Yu Y, Wang N, Hu R, Xiang F. Genome-wide identification of soybeantranscription factors in response to salt stress., 2016, 5(1): 1-15.
[43] Dou L, Zhang X, Pang C, Song M, Wei H, Fan S, Yu S. Genome-wide analysis of thegene family in cotton., 2014, 289(6): 1103-1121.
[44] Tang J, Wang F, Hou X L, Wang Z, Huang Z N. Genome-wide fractionation and Identification oftranscription factors in Chinese cabbage (ssp.) reveals collinearity and their expression patterns under abiotic and biotic stresses., 2013, 32(4): 1-15.
[45] Song H, Wang P, Lin J Y, Zhao C, Bi Y, Wang X. Genome-wide identification and characterization ofgene family in peanut., 2016, 7(1): 1-16.
[46] Huang S, Gao Y, Liu J, Peng X, Niu X, Fei Z, Cao S, Liu Y. Genome-wide analysis oftranscription factors in., 2012, 287(6): 495-513.
[47] Chen M, Tan Q, Sun M, Li D, Fu X, Chen X, Xiao W, Li L, Gao D. Genome-wide identification offamily genes in peach and analysis ofexpression during bud dormancy., 2016, 291(3): 1319-1332.
[48] Zhou Q Y, Tian A G, Zou H F, Xie Z M, Lei G, Huang J, Wang C M, Wang H W, Zhang J S, Chen S Y. Soybean-type transcription factor genes,,, and, confer differential tolerance to abiotic stresses in transgenicplants., 2008, 6(5): 486-503.
[49] Van verk M C, Pappaioannou D, Neeleman L, Bol J F, Linthorst H J. A noveltranscription factor is required for induction ofgene expression by salicylic acid and bacterial elicitors., 2008, 146(4): 1983-1995.
[50] Li C, Li D, Shao F, Lu S. Molecular cloning and expression analysis oftranscription factor genes in., 2015, 16(1): 1-21.
[51] Dai X., atranscription factor, modulates tolerance to phosphate starvation in rice., 2016, 67(3): 823-826.
[52] Su T, Xu Q, Zhang F C, Chen Y, Li L Q, Wu W H, Chen Y F.modulates phosphate homeostasis through regulating phosphate translocation and acquisition in., 2015, 167(4): 1579-1591.
[53] Wang H, Xu Q, Kong Y H, Chen Y, Duan J Y, Wu W H, Chen Y F.transcription factor activatesexpression in response to phosphate starvation., 2014, 164(4): 2020-2029.
[54] Wu H, Ni Z, Yao Y, Guo G, Sun Q. Cloning and expression profiles of 15 genes encodingtranscription factor in wheat (L.)., 2008, 18(6): 697-705.
[55] QIU Y P, JING S J, FU J, LI L, YU D Q. Cloning and analysis of expression profile of 13genes in rice., 2004, 49(20): 2159-2168.
[56] 黄胜雄, 刘永胜. 土豆转录因子家族的生物信息学分析. 应用与环境生物学报, 2013, 19(2): 205-214.Huang S X, Liu Y S. Genome-wide analysis oftranscription factors in., 2013, 19(2): 205-214. (in Chinese)
(责任编辑 李莉,岳梅)
Genome-wide Identification and Expression Analysis of WRKY Transcription Factor under Abiotic Stress in
KONG WeiLong1, YU Kun2, DAN NaiZhen1, YANG ShaoZong1, Bao ManZhu1,HUANG XiangRong1, FU XiaoPeng1
(1College of Horticulture and Forestry Sciences, Huazhong Agricultural University/Key Laboratory of Horticultural Plant Biology, Ministry of Education, Wuhan 430070;2College of Agronomy, Shihezi University/Key Laboratory of Oasis Ecological Agriculture, Xinjiang Production and Construction Group, Shihezi 832003, Xinjiang)
【Objective】WRKY transcription factor is a kind of transcription factor which plays an important role in plant response to biotic, abiotic stress, plant growth and development.Based on the sugar beet genome information, the WRKY family genes () were identified, tissue-specific expression profiles and expression pattern under salt and heat stresses were analyzed. The aim of the study is to provide reference for thegene function research, and lay a foundation for ornamental beet () and other Dianthus ornamental plant genetic engineering research. 【Method】 A total of 75WRKY proteins were used as
, according to WRKY conserved protein sequence (PF03106), thegenes ofwere identified by hmm and BLAST homology searches, and the chromosome location, phylogeny, gene structure, conserved domain, conserved element were also analyzed by MapInspect, GSDS2.0, MEGA5.0, DNAMAN5.0, WebLogo 3 and MEME bioinformatics tools. The specificity ofgenes expression and expression pattern under salt stress and heat stress ofwere analyzed by RNA-seq and qRT-PCR analysis. 【Result】gene family contained 40 members, 39 were unevenly distributed on 9 chromosomes, 1 on the random fragment. According to the WRKY conserved domain features and the evolution analysis with, 40 members were divided into three classes: Ⅰ, Ⅱ, and Ⅲ. classⅠhad 9 members, class Ⅱ had 26 members and class Ⅲ had 5 members. According to the evolutionary relationship, class Ⅱ further divided into five subclass: Ⅱa (1), Ⅱb (4), Ⅱc (9), Ⅱd (5) and Ⅱe (7). Genetic structure analysis showed that exon and intron number ofgenes had high variability (2-7 exons), even within the same subgroup. Conserved element analysis showed that within the same class or subclass had the same conserved elements. WRKY conserved domain analysis revealed two mutations in the WRKY domain: WRKYGKK and WRKYGEK. Eachwas expressed in at least two tissues, 30were expressed in leaf, 40were expressed in inflorescence, 36were expressed in young leaf, 38were expressed in root, 39were expressed in seedling, and 36were expressed in seed. The expression levels ofgenes were also different. Allgenes were divided into two types: low expression genes and high expression genes.Such as:,,,,,,,,,andwere highly expressed in all tissues;,,,,andwere low expressed in all tissues.,,,,andgenes were up-regulated under heat stress, and,,,andwere up-regulated under salt stress. In addition,had a significant response to both heat and salt stress. 【Conclusion】WRKY proteins are highly conserved, the length of the gene sequence and the number of introns varied widely, allgenes showed a variety of expression patterns in different tissues, somegenes responded to heat or salt stress, which play an important role in stress physiological regulation.
; WRKY transcription factor; bioinformatics; stress; expression analysis
2017-02-23;接受日期:2017-04-25
国家自然科学基金(31000918)、中央高校基本科研业务费专项(2662015PY052,2662016PY041)
孔维龙,E-mail:Asuraprince@126.com。通信作者傅小鹏,E-mail:fuxiaopeng@mail.hzau.edu.cn
猜你喜欢
中国畜牧杂志(2022年4期)2022-04-15
草业学报(2022年3期)2022-03-26
昆明医科大学学报(2022年1期)2022-02-28
小哥白尼(趣味科学)(2021年12期)2021-03-16
悦游 Condé Nast Traveler(2021年4期)2021-01-13
实用临床医药杂志(2021年13期)2021-01-10
中国实验诊断学(2017年5期)2017-06-05
中国糖料(2016年1期)2016-12-01
中国糖料(2016年1期)2016-12-01
中华胰腺病杂志(2015年5期)2015-12-08
