
油菜株高QTL定位、整合和候选基因鉴定
2017-09-26 08:02张江江詹杰鹏刘清云师家勤王新发刘贵华王汉中
中国农业科学 2017年17期
张江江,詹杰鹏,刘清云,师家勤,王新发,刘贵华,王汉中
油菜株高QTL定位、整合和候选基因鉴定
张江江1,詹杰鹏1,刘清云2,师家勤1,王新发1,刘贵华1,王汉中1
(1中国农业科学院油料作物研究所,武汉430062;2浠水县农业局油料作物推广站,湖北黄冈438200)
【目的】通过对油菜株高进行多环境QTL定位并与已报道的油菜株高QTL和植物株高基因分别进行整合和比对分析,揭示油菜株高的遗传结构和候选基因并为其分子改良提供依据。【方法】以油菜优良品种中双11(测序)和No.73290(重测序)衍生的含184个单株的BnaZNF2群体为试验材料。首先,对BnaZNF2群体进行基因型分析,利用Joinmap 4.0软件构建了一张含803个分子标记的高密度遗传图谱。其次,对F2:3和F2:4家系进行连续两年(2010—2011)两点(武汉和西宁)田间试验和表型鉴定。然后,利用BnaZNF2群体的基因型数据和F2:3以及F2:4家系的株高表型数据,采用WinQTLCart 2.5软件的复合区间作图法进行QTL检测。最后,利用元分析的方法采用BioMercator软件对不同环境中检测到的株高QTL进行整合。【结果】对两年两点环境下分别检测到的株高QTL进行整合总共得到5个株高QTL的位点:、、、和,分布于A2、C2和C3染色体上,解释2.6%—55.6%的表型方差。其中,和只在武汉检测到,而、和只在西宁检测到。位于C2连锁群的主效QTL-只在西宁被重复检测到,而且LOD值、加性效应和贡献率(分别为23.4、-16.0和55.6%)均高于前人报道,是目前发现的效应最大的一个油菜株高QTL。基于油菜基因组物理图谱对本研究和已报道的油菜株高QTL和植物株高基因分别进行整合和比对分析,获得了一个由183个QTL和287个候选基因组成的相对完整的油菜株高遗传结构图。其中,有18个株高QTL簇能在不同研究中被共同检测到,分布在A1、A2、A3、A6、A7、A9、C6和C7染色体上。另外,本研究定位到的5个油菜株高QTL的物理位置和已报道的油菜株高QTL均不重叠,因而是新的株高QTL位点。其中,、和物理区间内总共找到了15个株高同源基因,而11个在2个亲本中存在序列变异,被选作候选基因进行进一步研究。【结论】QTL定位和整合获得5个油菜株高QTL,均为首次报道而且都只在武汉或西宁被检测到。其中位于C2连锁群的主效QTL效应值超过以往报道,表现出极强的QTL与环境的互作。通过与已报道的油菜株高QTL和植物株高基因分别进行整合和比对分析,较为全面地揭示了油菜株高的遗传结构和候选基因,生物信息学分析还鉴定到11个位于本研究定位到的3个株高QTL区间内的候选基因。
甘蓝型油菜;株高;遗传结构;QTL;候选基因;QTL与环境的互作
0 引言
【研究意义】油菜是中国第一大油料作物[1],也是中国唯一的冬季油料作物[2],近年来,种植面积和总产均占全世界的20%左右(http://apps.fas.usda. gov/psdonline/)。菜籽油是世界植物油第二大来源,同时也是中国食用植物油第一大来源,占中国国产油料作物产油量的55%以上,在国家食用油供给安全战略中地位十分重要[3-4]。然而,由于单产和机械化水平低共同造成的生产效益低下是制约油菜产业发展的主要因素[3]。从作物品种的演变历程可以看出,品种产量提高和种植方式升级的背后往往伴随着株型的选择和优化。株高是油菜株型的主要决定因素之一[5-6],不但与油菜收获指数以及产量密切相关,而且也是影响抗倒伏能力和机械化收获特性的一个重要因素[3,7]。因此,油菜株高遗传结构的解析,对于培育油菜理想株型品种具有重要的理论与现实意义。【前人研究进展】油菜株高是一个典型的数量性状,表型连续分布且容易受到环境条件的影响[3]。近十年来,利用连锁[8-13]或关联分析[5,14-21]的方法,在油菜中已经定位到200多个株高QTL。这些株高QTL在油菜所有19个连锁群上都有分布,其中绝大部分贡献率都比较低,而只在A2、A3、C2和C6连锁群上发现了少数几个效应较大的QTL。Shi等[15]用源自Tapidor和宁油7号的202个双单倍体及其衍生的“重构F2”群体,在10个环境中共定位到44个株高QTL,分布于A1-3、A5-10、C3、C6、C7和C9染色体上。LI等[12]用472个油菜核心种质和60K油菜SNP芯片进行全基因组关联分析,在3个环境中共检测到8个株高QTL,分布于A3、A5、A7和C7染色体上。Wang等[13]用显性矮杆突变体(源自油菜纯系NJ7982的EMS诱变)和中双11衍生的回交分离群体将该位点精细定位到A9染色体152 kb的区段,并从14个注释基因中鉴定了7个候选基因。Liu等[22]克隆了油菜A6染色体上一个半显性矮杆突变体基因(源自92-B10双单倍体的EMS诱变),该基因()编码DELLA蛋白作为赤霉素信号的受体。虽然油菜株高基因的克隆尚处于起步阶段,但在模式植物拟南芥和主要农作物中已经鉴定了一大批调控株高的基因[23-24],大多与赤霉素、油菜素内酯和生长素等植物激素的合成或信号转导途径有关。【本研究切入点】因为不同研究者进行油菜株高QTL定位的遗传图谱或标记系统不同,很难准确判断这些QTL之间的位置关系进而将它们整合,因此无法揭示一个相对完整的油菜株高的遗传结构图。另外,检测到的油菜株高QTL效应偏小难以被精细定位并克隆,而植物中揭示的株高基因及其调控途径可以为油菜株高的基因克隆和机理研究提供借鉴和参考。【拟解决的关键问题】基于油菜测序品种中双11和No.73290构建的BnaZNF2群体,对油菜株高性状进行多年多点QTL定位,同时利用已发表的油菜基因组物理图谱和已报道的株高QTL进行整合和比较,最后利用植物中已克隆的株高基因进行同源比对并筛选QTL区间内的候选基因,系统地揭示油菜株高的遗传结构及候选基因,为油菜株高的遗传研究和分子改良打下基础。
1 材料与方法
2009年10月至2010年5月、2010年10月至2011年5月在武汉中国农业科学院油料作物研究所阳逻综合试验基地完成了BnaZNF2:3家系群体种植及田间考察工作。2011年4—8月在西宁青海大学农场完成了BnaZNF2:3和BnaZNF2:4家系群体的田间种植和表型鉴定工作。在这3个家系群体的基础上于2016年对数据进行了QTL扫描定位和整合工作。
1.1 试验材料
用来进行株高QTL定位的群体名为BnaZNF2[25],来源于油菜测序品种中双11(https://www.ncbi.nlm.nih. gov/genome/genomes/203?)和No.73290[26]。F2:3家系群体的种子由184个F2单株分别套袋自交所得;而F2:4家系群体的种子则由F2:3家系各单株分别套袋自交所得种子按数目等量混合而成。
1.2 田间试验
田间试验采用完全随机区组设计,3次重复。每小区种两行,行距33.3 cm,单株间平均间距16.7 cm。待油菜成熟时,每个小区随机挑选10个有代表性的单株手工收获晾干。
1.3 性状考察和数据分析
株高考察参考油菜品种审定标准[27],自子叶节至全株最高部分长度,以“cm”表示。用SAS V8软件的PROC ANOVA程序对几个环境的株高表型数据进行方差分析,利用遗传方差模型对遗传力进行估算。广义遗传力的计算公式:2=σg2/(σg2+σge2/n+σe2/nr)。其中,σg2、σge2和σe2分别代表基因型方差、基因型和环境互作方差以及误差项;n和r分别代表环境数和重复数。
1.4 QTL定位
油菜分子育种课题组之前利用BnaZNF2群体构建的遗传连锁图谱进行QTL定位[25]。该连锁图谱包含803个标记,19个连锁群,图谱总长1 763.2 cM。利用软件WinQTLCart 2.5软件[28]和复合区间作图(composite interval mapping)的方法[29-30]进行QTL扫描。LOD阈值用1 000次重复排列测验确定。基本参数设置:步长=1 cM;窗口大小=10 cM;控制标记数=5。用=0.05水平下的LOD阈值来确定显著的QTL,为了避免漏掉微效QTL,那些可重复的=0.1水平下的QTL也被承认[6]。按照McCouch等[31]的方法对检测到的QTL命名,以“”加上性状再加染色体编号表示,字体为斜体。采用元分析[32]的方法对不同环境(地点×年份组合)中检测到的QTL进行整合。
1.5 候选基因的鉴定
从已发表的文章中[6]收集植物中控制株高的基因约200个[33-49]。利用上述收集到的株高基因序列和油菜基因组注释基因序列进行Blast比对(参数设置为:e-10),确认它们在油菜基因组中的同源基因。
2 结果
2.1 亲本和群体株高表型和遗传力分析
亲本中双11和No.73290的株高差别不大,两者间的差异只在西宁2011年达到了显著水平(图1和表1)。但是,无论是BnaZNF2:3还是BnaZNF2:4群体株高在各个环境下都表现出广泛的变异和超亲分离,表明株高增效基因在2个亲本中都有分布。而且,2个群体在各个环境下的株高都呈正态或近似正态分布,符合数量性状的典型特征,表明该群体适合用来进行QTL定位。
通过对上述几个环境下的株高进行方差分析(表2),结果表明,无论是基因型、环境还是它们之间的互作对株高的效应都达到了极其显著的水平。通过方差组分估计出来的株高遗传力为0.54。
2.2 多环境下株高QTL定位
用软件WinQTLCart 2.5对武汉、西宁两年4个环境下的株高数据分别进行QTL扫描。检测到的单环境下的QTL经元分析整合后得到5个QTL(、、、和)分别位于A2、C2和C3连锁群。这些QTL的LOD值在3.09—23.4,贡献率在2.6%—55.6%(表3)。其中,和只在武汉检测到,而、和只在西宁检测到,说明环境对株高QTL的表达有很大影响。另外和的加性效应为正,说明这2个QTL的增效等位基因来自中双11,而、和加性效应是负的,说明这3个QTL的增效等位基因来自No.73290。

表1 2个亲本和群体在3个环境下的株高表型
在西宁2011年2个群体都重复检测到了一个效应值很高的主效QTL,其LOD值、加性效应和贡献率(分别为23.4、-16.0和55.6%)均高于前人报道,是目前为止发现的效应最大的一个油菜株高QTL。这个主效QTL只在西宁重复检测到,而在武汉2年均检测不到,呈现出很强的与环境的互作(图2)。武汉和西宁在油菜种植期内的主要环境差别是日照长度和温度,而有研究表明二者会影响植物株高[21,49]。因此,推测很有可能和日照长短或温度有关。
2.3 和已报道的油菜株高QTL的整合与比较
油菜中已经有10多篇文献报道了株高QTL的连锁或关联定位结果,共定位243个,分布在油菜所有19个连锁群,其中,以A2、A3、A6、A7连锁群分布较为集中。
为了进一步准确地比较这些QTL的位置,利用其连锁/关联的分子标记引物/探针序列和已发表的油菜基因组参考序列进行比对分析,确定了其中183个QTL的物理位置(图3)。同时,也确定了定位的5个株高QTL的物理位置。通过比较发现,定位的5个QTL与前人报道的油菜株高QTL均没有重叠,因而是新的油菜株高QTL位点。
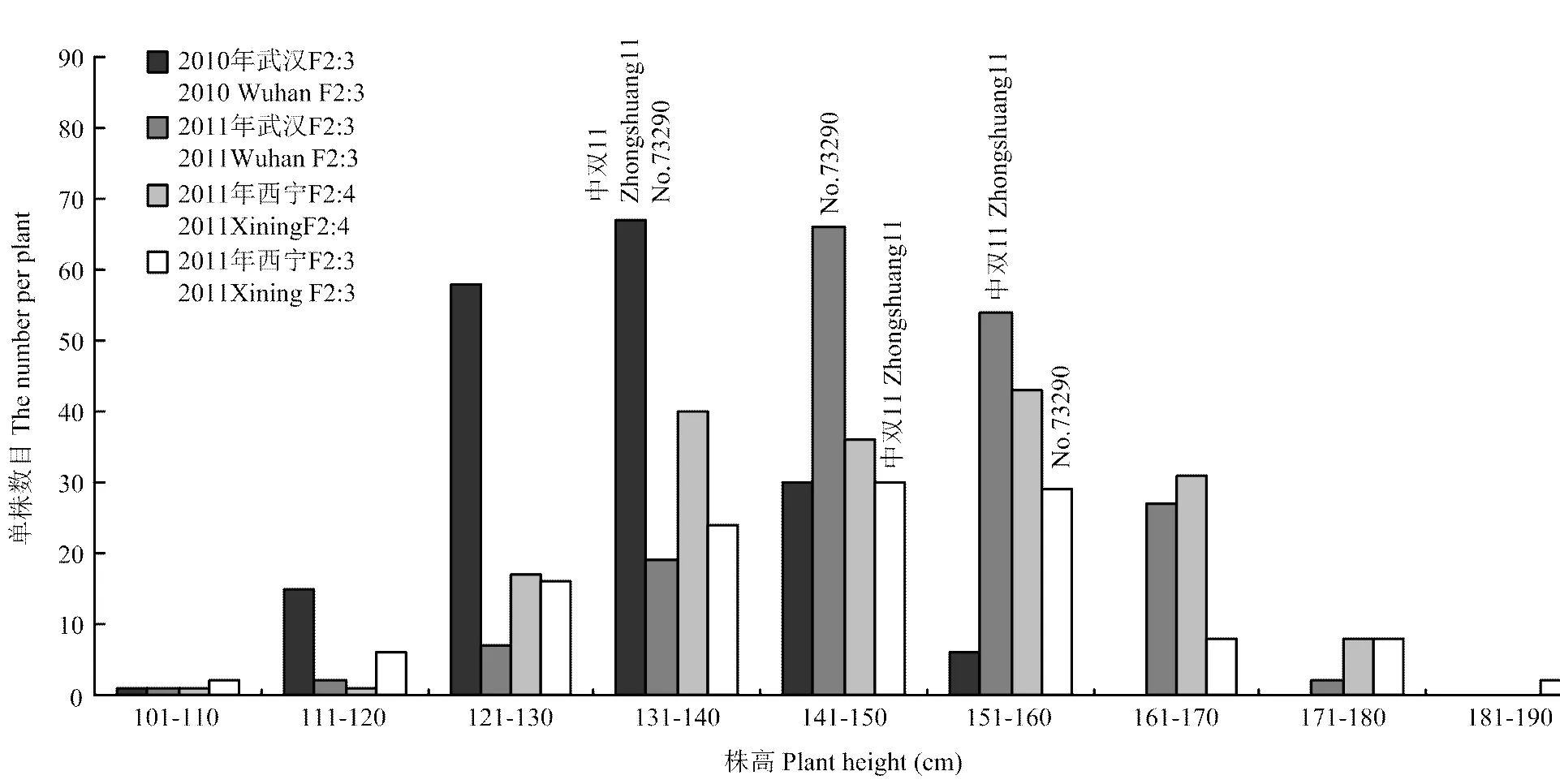
图1 BnaZNF2:3和BnaZNF2:4群体两年两点株高的频率分布
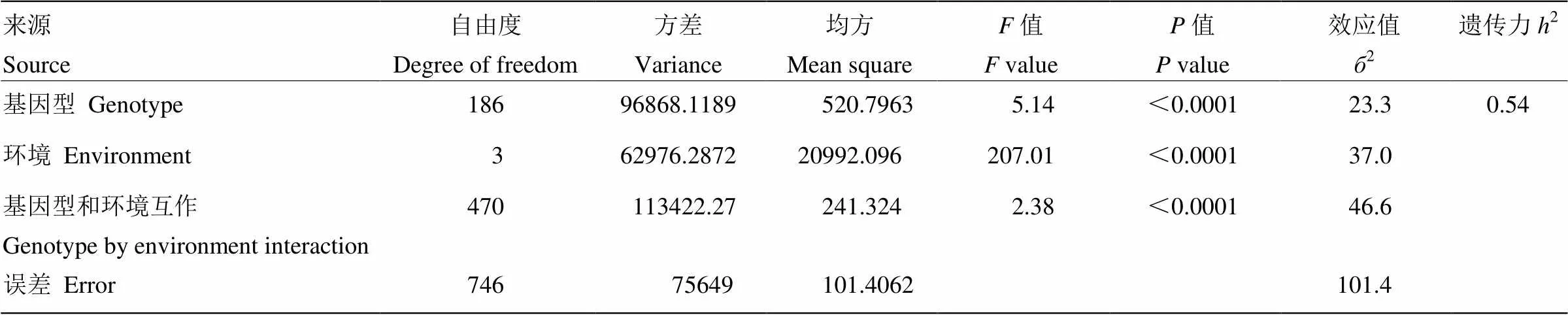
表2 株高表型方差分析和遗传力估算
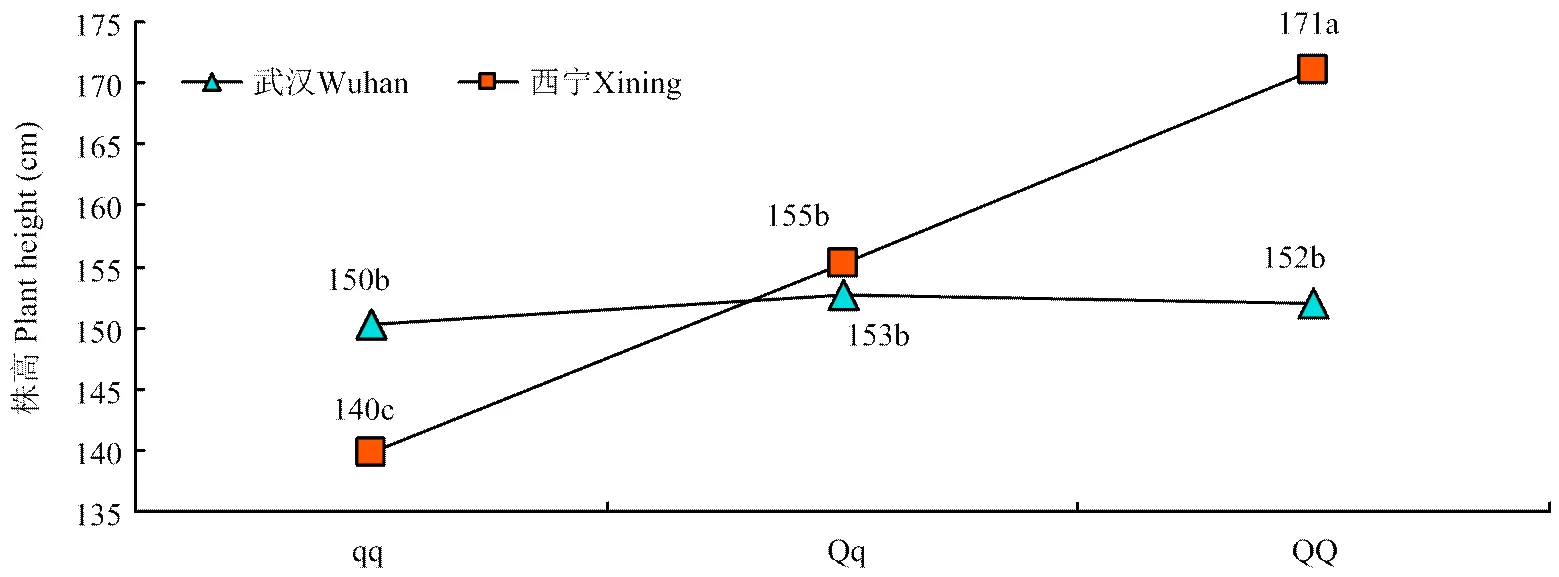
图2 qPH.C2-1与环境的互作
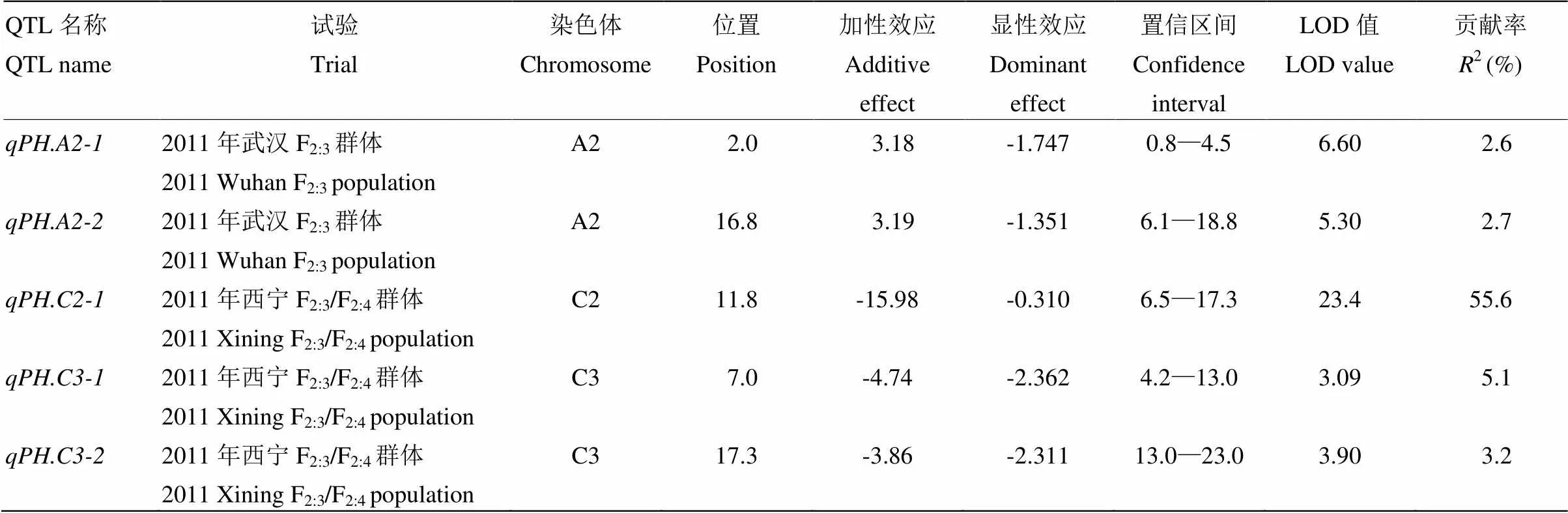
表3 武汉和西宁环境下检测出的株高QTL
另外,综合比较和前人研究中报道的株高QTL的物理位置,发现18个重叠的QTL簇,它们分布在A1、A2、A3、A6、A7、A9、C6和C7染色体上(表4)。这些能在不同的群体或环境中共同检测到的QTL可信度高、稳定性好,应成为油菜株高分子标记辅助选择研究的重点。
2.4 候选基因鉴定
通过广泛查阅文献,收集到223个控制植物株高的基因,用它们的序列和油菜基因序列进行Blast分析,发现它们在油菜参考基因组中共有727个同源基因(图3)。其中,287个位于上述QTL的物理区间内,可以作为这些株高QTL的候选基因。本研究定位到的、和物理区间内分别找到了4、7和4个株高同源基因(表5)。其中,有11个株高同源基因在亲本中存在序列差异,可以作为候选基因进行后续研究。而和物理区间内没有找到株高同源基因,说明它们很可能是由新的株高基因控制的。

表4 不同研究检测到的共同的油菜株高QTL
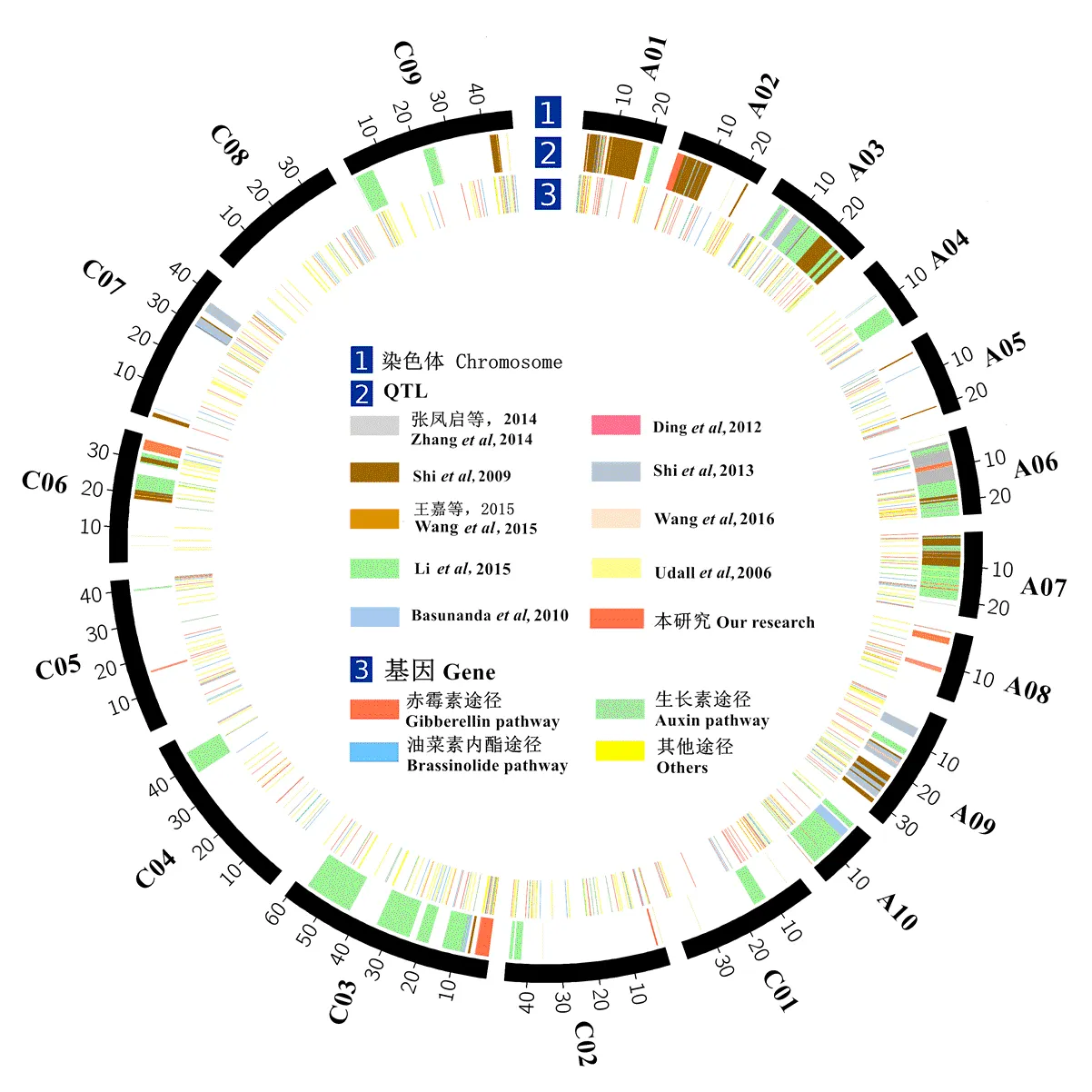
图3 油菜株高QTL和同源株高基因在物理图谱上的分布
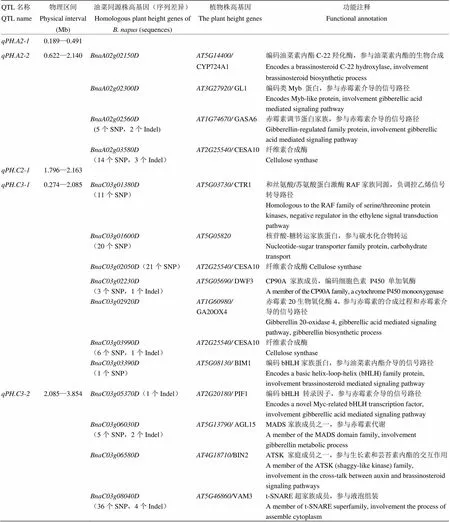
表5 株高QTL候选基因鉴定
3 讨论
虽然前人利用连锁或关联分析的方法在油菜中已定位了200多个株高QTL,但由于所用的遗传图谱和标记系统不同难以将它们整合,因此尚未获得一个相对完整的油菜株高的遗传结构图。基于已发表的油菜参考基因组序列[50],将这些油菜株高QTL整合在同一个物理图谱上,从而获得了首张油菜株高遗传结构图。这些QTL分布在油菜所有19个连锁群,其中绝大部分贡献率都在10%以下,而只有11个效应较大,可以作为后续精细定位和基因克隆的目标。另外,本研究还发现了在不同研究中共同检测到的18个油菜株高QTL簇,可以作为分子标记辅助选择的靶点。这些研究结果说明油菜株高是一个由众多基因控制的数量性状,具有非常复杂的遗传基础。
本研究定位了5个株高QTL,都与已报道的株高QTL位置不重叠,应该代表新的油菜株高QTL位点。其中,只在西宁(春油菜生态区)而不是武汉(半冬性油菜生态区)被重复检测到,可以作为研究QTL和环境互作效应的范例,这也说明环境对株高QTL表达有很大影响。另外,的贡献率和加性效应都高于前人的报道,可以作为下一步基因克隆的首选目标。本研究定位的这5个株高QTL物理区间(范围:0.302—1.811 Mb;均值:1.153 Mb)略小于前人的报道,这可能跟它们都位于染色体较为末端的位置有关,因为这些区域的重组更为频繁。
目前,油菜株高遗传研究整体上还处于QTL初步定位阶段,尚无油菜株高QTL被精细定位,更没有被克隆的报道。这很可能是因为已报道的株高QTL效应都小,而且株高受环境条件的影响较大而表型难以准确鉴定,因此,难以进行精细定位和克隆。因此,很多研究者转而利用油菜株高突变体进行研究,精细定位或克隆了少数几个株高基因[10,50]。而本研究发现的油菜株高主效QTL是目前为止效应最大的,完全具备进一步精细定位和克隆的基础,从而为油菜株高的遗传改良打下理论基础并提供技术支撑。
4 结论
QTL定位和整合获得了5个油菜株高QTL,均为首次报道而且都只在武汉或西宁被检测到。其中位于C2连锁群的主效QTL效应值超过以往报道,表现出极强的QTL与环境的互作。通过与已报道的油菜株高QTL和植物株高基因分别进行整合和比对分析,较为全面地揭示了油菜株高的遗传结构和候选基因。另外,鉴定到11个位于本研究定位到的3个株高QTL区间内的候选基因。
References
[1] Hu Q, Hua W, Yin Y, Zhang X K, Liu L J, Shi J Q, Zhao Y G, Qin L, Chen C, Wang H Z. Rapeseed research and production in China., 2017, 5(2): 127-135.
[2] James C. Global review of commercialized transgenic crops., 2003, 84: 303-309.
[3] 王汉中, 殷艳. 我国油料产业形势分析与发展对策建议. 中国油料作物学报, 2014, 36(3): 414-421.
Wang H Z, Yin Y. Analysis and strategy for oil crop industry in China., 2014, 36(3): 414-421. (in Chinese)
[4] 沈金雄, 傅廷栋. 我国油菜生产、改良与食用油供给安全. 中国农业科技导报, 2013, 13(1): 1-8.
Shen J X, Fu T D. Rapeseed production, improvement and edible oil supply in China., 2013, 13(1): 1-8. (in Chinese)
[5] Thuiling N. Application of the ideotype concept in breeding for higher yield in the oilseed brassicas., 1991, 26(2): 201-219.
[6] 易斌, 陈伟, 马朝芝, 傅廷栋, 涂金星. 甘蓝型油菜产量及相关性状的QTL分析. 作物学报, 2006, 32(5): 676-682.
Yi B, Chen W, Ma C Z, Fu T D, Tu J X. Mapping of quantitative trait loci for yield and yield components inL.., 2006, 32(5): 676-682. (in Chinese)
[7] 王汉中. 我国油菜产业发展的历史回顾与展望. 中国油料作物学报, 2010, 32(2): 300-302.
Wang H Z. Review and future development of rapeseed industry in China., 2010, 32(2): 300-302. (in Chinese)
[8] Wang Y K, He J B, Yang L, Wang Y, Chen W J, Wan S B, Chu P, Guan R Z. Fine mapping of a major locus controlling plant height using a high‑density single‑nucleotide polymorphism map in., 2016, 129(8): 1479-1491.
[9] 张凤启, 刘越英, 程晓辉, 童超波, 董彩华, 唐敏强, 黄军艳, 刘胜毅. 利用高密度SNP标记定位甘蓝型油菜株高QTL. 中国油料作物学报, 2014, 36(6): 695-700.
Zhang F Q,Liu Y Y, Cheng X H, Tong C B, dong c h, Tang M Q, Huang J Y, Liu S Y. QTL mapping of plant height using high density SNP markers in., 2014, 36(6): 695-700. (in Chinese)
[10] Ding G D, Zhao Z K, Liao Y, Hu Y F, Shi L, Long Y, Xu F S. Quantitative trait loci for seed yield and yield-related traits, and their responses to reduced phosphorus supply in., 2012, 109(4): 747-759.
[11] Quijada P A, Udall A J, Lambert B, Osborn T C. Quantitative trait analysis of seed yield and other complex traits in hybrid spring rapeseed (L.): 1. identification of genomic regions from winter germplasm., 2006, 113(3): 549-561.
[12] Li F, Chen B Y, Xu K, Gao G Z, Yan G X, Qiao J W, Li J, Li H, Li L X, Xiao X, Zhang T Y, Takeshi N, Wu X M. A genome-wide association study of plant height and primary branch number in rapeseed ()., 2016, 242: 169-177.
[13] Wang X D, Wang H, Long Y, Liu L Z, Zhao Y J, Tian J H, Zhao W G, Li B J, Chen L, Chao H B, Li M T. Dynamic and comparative QTL analysis for plant height in different developmental stages ofL.., 2015, 128(6): 1175-1192.
[14] Cai D F, Xiao Y J, Yang W, Ye W, Wang B, Muhammad Y, Wu J S, Liu K D. Association mapping of six yield‑related traits in rapeseed (L.)., 2014, 127: 85-96.
[15] Shi T X, Li R Y, Zhao Z K, Ding G D, Long Y, Meng J L, Xu F S, Shi L. QTL for yield traits and their association with functional genes in response to phosphorus deficiency in., 2013, 8(1): e54559.
[16] Basunanda P, Radoev M, Ecke W, Friedt W, Becker H C, Snowdon R J. Comparative mapping of quantitative trait loci involved in heterosis for seedling and yield traits in oilseed rape (L.)., 2010, 120(2): 271-281.
[17] Valiollah R. Combining ability analysis of plant height and yield components in spring type of rapeseed varieties (L.) using line × tester analysis., 2012, 2(1): 58-62.
[18] Mladen R, Heiko C B, Wolfgang E. Genetic analysis of heterosis for yield and yield components in rapeseed (L.) by quantitative trait locus mapping., 2008, 179(3): 1547-1558.
[19] Mei D S, Wang H Z, Hu Q, Li Y D, Xu Y S, Li Y C. QTL analysis on plant height and flowering time in., 2009, 128(5): 458-465.
[20] Zhang S F, Fu T D, Zhu J C, Wang J P, Wen Y C, Ma C Z, Jiang Y Z. QTL mapping and epistasis analysis for plant height and height to the first branch in rapeseed (L.)., 2007, 2: 232-235.
[21] 王嘉, 荆凌云, 荐红举, 曲存民, 谌利, 李加纳, 刘列钊. 甘蓝型油菜株高、第一分枝高和分枝数的QTL检测及候选基因的筛选. 作物学报, 2015, 41(7): 1027-1038.
Wang J, Jing L Y, Jian H J, Qu C M, Chen L, Li J N, Liu L Z. Quantitative trait loci mapping for plant height, the first branch height, and branch number and possible candidate genes screening inL.., 2015, 41(7): 1027-1038. (in Chinese)
[22] Liu C, Wang J L, Huang T D, Wang F, Yuan F, Cheng X M, Zhang Y, Shi S W, Wu J S, Liu K D. A missense mutation in the VHYNP motif of a DELLA protein causes a semi-dwarf mutant phenotype in., 2010, 121(2): 249-258.
[23] Ma X S, Feng F J, Wei H B, Mei H W, Xu K, Chen S J, Li T F, Liang X H, Liu H Y, Luo L J. Genome-wide association study for plant height and grain yield in rice under contrasting moisture regimes., 2016, 144(6): 651-664.
[24] Shi J Q, Li R Y, Qiu D, Jiang C C, Long Y, Morgan C, Bancroft I, Zhao J Y, Meng J L. Unraveling the complex trait of crop yield with quantitative trait loci mapping in., 2009, 182(3): 851-861.
[25] Shi J Q, Zhan J P, Yang Y H, Ye J, Huang S M, Li R Y, Wang X F, Liu G H, Wang H Z. Linkage and regional association analysis reveal two new tightly-linked major-QTLs for pod number and seed number per pod in rapeseed (L.)., 2015, 5: 14481.
[26] Huang S M, Deng L B, Guan M, Li J N, Lu K, Wang H Z, Fu D H, Annaliese S M, Liu S Y, Hua W. Identification of genome-wide single nucleotide polymorphisms in allopolyploid crop., 2013, 14(1): 717.
[27] 张芳. 我国油菜品种审定管理与育种趋势研究[D]. 北京: 中国农业科学院, 2012.
Zhang F. Rape cultivar breeding and management trends[D]. Beijing: Chinese Academy of Agricultural Sciences, 2012. (in Chinese)
[28] Wang S, Basten C J, Gaffney P, Zeng Z B. Windows QTL Cartographer version 2.5. North Carolina State University. Bioinformatics Research Center, Raleigh, 2001. http://statgen.ncsu.edu/ qtlcart/WQTLCart,htm.
[29] Zeng Z B. Theoretical basis for separation of multiple linked gene effects in mapping quantitative trait loci., 1993, 90(23): 10972-10976.
[30] Zeng Z B. Precision mapping of quantitative trait loci., 1994, 136(4): 1457-1468.
[31] McCouch S R, Cho Y G, Yano M, Paul E, Blinstrub M, Morishima H, Kinoshita T. Report on QTL nomenclature, 1997, 14(11): 11-13.
[32] Goffinet B, Gerber S. Quantitative trait loci: a meta-analysis., 2000, 155(1): 463-473 .
[33] Kujur A, Upadhyaya H D, Bajaj D, Gowda C L L, Sharma S, Tyagi A K, Parida S K. Identification of candidate genes and natural allelic variants for QTLs governing plant height in chickpea., 2016, 6: 27968.
[34] Teng F, Zhai L H, Liu R X, Bai W, Wang L Q, Huo D G, Tao Y S, Zheng Y L, Zhang Z X. ZmGA3ox2, a candidate gene for a major QTL,, for plant height in maize., 2013, 73(3): 405-416.
[35] Weng J, Xie C, Hao Z, Wang J, Liu C, Li M, Zhang D, Bai L, Zhang S, Li X. Genome-wide association study identifies candidate genes that affect plant height in chinese elite maize (L.) inbred lines., 2011, 6(12): e29229.
[36] María L R, Emiliano A, Mariano B, Carlos A S. Phenotypic characterization, genetic mapping and candidate gene analysis of a source conferring reduced plant height in sunflower., 2013, 126(1): 251-263.
[37] Li X P, Zhou Z J, Ding J Q, Wu Y B, Zhou B, Wang R X, Ma J L, Wang S W, Zhang X C, Xia Z L, Chen J F, Wu J Y. Combined linkage and association mapping reveals QTL and candidate genes for plant and ear height in maize., 2016, 7: 833.
[38] Ma X S, Feng F J, Wei H B, Mei H W, Xu K, Chen S J, Li T F, Liang X H ,Liu H Y, Luo L J. Genome-wide association study for plant height and grain yield in rice under contrasting moisture regimes., 2016, 7: 1801.
[39] Zanke C D, Ling J, Plieske J, Kollers S, Ebmeyer E, Korzun V, Argillier O, Stiewe G, Hinze M, Neumann F, Eichhorn A, Polley A, Jaenecke C, Ganal M W, Röder M S. Analysis of main effect QTL for thousand grain weight in european winter wheat (L.) by genome-wide association mapping., 2016, 6: 644.
[40] Lim J H, Yang H J, Jung K H, Yoo S C, Paek N C.quantitative trait locus mapping and candidate gene analysis for plant architecture traits using whole genome re-sequencing in rice, 2014, 37(2): 149-160.
[41] Wang Y J, Xu J, Deng D X, Ding H D, Bian Y L, Yin Z T, Wu Y R, Zhou B, Zhao Y. A comprehensive meta-analysis of plant morphology, yield, stay-green, and virus disease resistance QTL in maize (L.)., 2016, 243: 459-471.
[42] Peng Y L, Gao Z Y, Zhang B, Liu C L, Xu J, Ruan B P, Hu J, Dong G J, Guo L B, Liang G H, Qian Q. Fine mapping and candidate gene analysis of a major QTL for panicle structure in rice., 2014, 33: 1843-1850.
[43] Vemireddy L R, Noor S, Satyavathi V V, Srividhya A, Kaliappan A, Parimala SRN, Bharathi P M, Deborah D A, Rao K S, Shobharani N, Siddiq E A, Nagaraju J. Discovery and mapping of genomic regions governing economically important traits of Basmati rice, 2015, 15: 207.
[44] Mallikarjuna R K, Zhang Y S, Yu S B, Yang G Y, Yan W H, Xing Y Z. Candidacy of a chitin-inducible gibberellin-responsive gene for a major locus affecting plant height in rice that is closely linked to Green Revolution gene., 2011, 123: 705-714.
[45] Bensen R J, Johal G S, Crane V C, Tossberg J T, Schnable P S, Meeley R B, Briggs S P. Cloning and characterization of the maizegene., 1995, 7(1): 75-84.
[46] Fujioka S, Yamane H, Spray C R, Gaskin P, Macmillan J, Phinney B O, Takahashi N. Qualitative and quantitative analyses of gibberellins in vegetative shoots of normal,dwarf-1, dwarf-2, dwarf-3, and dwarf-5 seedlings ofL.,1988, 88(4): 1367-1372.
[47] Spray C R, Kobayashi M, Suzuki Y, Phinney B O, Gaskin P, MacMillan J. The dwarf-i (dl) mutant ofblocks three steps in the gibberellin-biosynthetic pathway.,1996, 93(19): 10515-10518
[48] Liu T, Zhang J, Wang M, Wang Z, Li G, Qu L, Wang G. Expression and functional analysis of ZmDWF4, an ortholog ofDWF4 from maize (L.)., 2007, 26(12): 2091-2099.
[49] Weng F J, Xie C X, Hao Z F, Wang J J, Liu C L, Li M S, Zhang D H, Bai L, Zhang S H, Li X H. Genome-wide association study identifies candidate genes that affect plant height in Chinese elite maize (L.) inbred lines., 2011, 6(12): e29229.
[50] Chalhoub B, Denoeud F, Liu S Y, Parkin I A, Tang H B, Wang X Y, Chiquet J, Belcram H, Tong C B, Samans B, Corréa M, Da S C, Just J, Falentin C, Koh C S, Le C I, Bernard M, Bento P, Noel B, Labadie K, Alberti A, Charles M, Arnaud D, Guo H, Daviaud C, Alamery S, Jabbari K, Zhao M X, Edger P P, Chelaifa H, Tack D, Lassalle G, Mestiri I, Schnel N, Paslier M C, Fan G G, Renault V, Bayer P E, Golicz A A, Manoli S, Lee T H, Thi V H, Chalabi S, Hu Q, Fan C C, Tollenaere R, Lu Y H, Battail C, Shen J X, Sidebottom C H, Wang X F, Canaguier A, Chauveau A, Bérard A, Deniot G, Guan M, Liu Z S, Sun F M, Lim Y P, Lyons E, Town C D, Bancroft I, Wang X W, Meng J L, Ma J X, Pires J C, King G J, Brunel D, Delourme R, Renard M, Aury J M, Adams K L, Batley J, Snowdon R J, Tost J, Edwards D, Zhou Y M, Hua W, Sharpe A G, Paterson A H, Guan C Y, Wincker P. Early allopolyploid evolution in the post-Neolithicoilseed genome.,2014, 345(6199): 950-953.
(责任编辑 李莉,岳梅)
QTL mapping and integration as well as candidate genes identification for plant height in rapeseed (L.)
ZHANG JiangJiang1, ZHAN JiePeng1, LIU QingYun2, SHI JiaQin1, WANG XinFa1,LIU GuiHua1, WANG HanZhong1
(1Oil Crops Research Institute, Chinese Academy of Agricultural Sciences, Wuhan 430062;2Oil Crops Extension Station of the Agricultural Bureau of Xishui County, Huanggang 438200, Hubei)
【Objective】In order to reveal the genetic architecture and candidate genes for plant height in rapeseed, QTLs were mapped in multiple environments and were integrated with previously reported plant height QTLs and then aligned with the plant height genes, which will provide a basis for the molecular improvement of plant height in rapeseed. 【Method】The BnaZNF2population of 184 individuals derived from the elite rapeseed cultivar Zhongshuang11 (de novo sequencing) and No.73290 (re-sequencing) was used as the experimental material. First, the BnaZNF2population was subjected to genotype analysis and a high-density linkage map of 803 molecular markers was constructed using Joinmap 4.0. Second, the F2:3and F2:4family of BnaZNF2population were planted and phenotyped at two locations (Wuhan and Xining) for successive two years (2010 and 2011). Then QTL mapping was conducted by the composite interval mapping method incorporated into WinQTLCart 2.5 software, using the genotype of BnaZNF2population and the plant height phenotype of its F2:3and F2:4family. 【Result】After integration of QTLs detected in two locations over two years, a total of 5 consensus QTLs (,,,,) were obtained, which were distributed on A2, C2 and C3 chromosomes and, explained 2.6%-55.6% of the phenotypic variance. A major QTL on the C2 chromosome,, was only detected repeatedly in Xining and its LOD value, additive effect and2(23.4, -16.0 and 55.6%, respectively) were largest among all of the reported plant height QTLs. Based on the physical map of rapeseed, all of the currently and previously reported plant height QTLs in rapeseed were integrated and then aligned with the plant height genes, which revealed a relatively completed genetic architecture map consisting of 183 QTLs in rapeseed and 287 candidate genes in rapeseed. Of these, a total of 18 QTL cluster were commonly detected in different studies, which were distributed on A1, A2, A3, A6, A7, A9, C6 and C7 chromosomes. In addition, the physical positions of the five QTL detected in the current study were all not overlapped with those of the previously detected plant height QTL, which should be novel. A total of 15 homologues of plant height genes were found within the physical intervals of,and, of which 11 homologues showed sequence variations between the two parents, which were chosen as the candidates for further study. 【Conclusion】QTL mapping and integration identified five QTL for plant height in rapeseed, which were all novel. The effect of the major QTL on the C2 chromosome was larger than those of the previously reported plant height QTL, which also showed the strong interaction with the environment. The integration of the reported plant height QTLs and the alignment with the plant height genes systematically revealed the genetic architecture and candidate genes for plant height in rapeseed. By bioinformatics analysis, a total of 11 candidates were identified within the physical intervals of three plant height QTLs detected in the current study.
L.; plant height; genetic architecture; QTL; candidate genes; QTL by environment interaction
2017-01-20;接受日期:2017-04-05
国家油菜产业技术体系(CARS-13)、中国农业科学院科技创新工程(CAAS-ASTIP-2013-OCRI)、国家公益性科研院所基本科研业务费(1610172017001)、湖北农业科技创新中心
张江江,E-mail:zhangjiangjiang6@163.com。通信作者师家勤,Tel:027-86711553;E-mail:shijiaqin@caas.cn
猜你喜欢
Chinese Physics B(2022年5期)2022-05-16
现代装饰(2020年7期)2020-07-27
农村百事通(2019年17期)2019-10-08
中国水土保持(2019年7期)2019-08-08
金桥(2018年7期)2018-09-25
NBA特刊(2018年7期)2018-06-08
现代农业科技(2017年1期)2017-03-06
江苏农业科学(2016年8期)2017-02-15
江苏农业科学(2016年8期)2017-02-15
汽车维护与修理(2015年6期)2015-02-28
