
黔产喜树叶的HPLC指纹图谱研究及聚类分析Δ
2017-09-07 00:34刘志海范红梅贵州省黔西南州食品药品检验检测中心贵州兴义562400
中国药房 2017年24期
刘志海,范红梅,邹 兰,隆 林,金 昭,余 兰(贵州省黔西南州食品药品检验检测中心,贵州兴义562400)
黔产喜树叶的HPLC指纹图谱研究及聚类分析Δ
刘志海*,范红梅,邹 兰,隆 林,金 昭,余 兰(贵州省黔西南州食品药品检验检测中心,贵州兴义562400)
目的:建立黔产喜树叶的高效液相色谱(HPLC)指纹图谱。方法:采用HPLC法。色谱柱为Gemini-NX C18,流动相为乙腈-0.2%磷酸溶液(梯度洗脱),流速为1.0 mL/min,检测波长为370 nm,柱温为30℃,进样量为10 μL。以山柰素为参照物,测定14批喜树叶药材的HPLC图谱,采用《中药色谱指纹图谱相似度评价系统》(2004 A)进行共有峰指认、相似度评价,并进行聚类分析。结果:14批喜树叶药材的HPLC图谱共有10个共有峰,13批喜树叶药材与对照图谱的相似度>0.90,1批与对照图谱相似度<0.90。喜树叶药材样品可归为3大类。结论:该研究所建指纹图谱可为黔产喜树叶的鉴别和质量评价提供参考。
喜树叶;指纹图谱;聚类分析;高效液相色谱法
喜树叶为蓝果树科喜树属植物喜树Camptotheca acuminata Decne的叶,喜树是我国特有树种,主要分布在长江流域及西南各省[1],多选其果实、根、根皮、树皮、叶入药。喜树叶具有清热解毒、祛风止痒之功效,主治痈疮疖肿、牛皮癣。据文献研究报道,喜树叶中含喜树碱、10-羟基喜树碱、槲皮素和山柰酚等有效成分[2]。喜树在贵州各地分布较为广泛,2003年版《贵州省中药材、民族药材质量标准》收载了喜树果的质量标准,但未收载喜树叶[3]。为全面提升我省中药材、民族药材质量标准,现开展对喜树叶药材质量标准研究工作。本研究参考文献[4],结合我省资源情况,收集了贵州8个地州市共计14批喜树叶药材样本,对其进行指纹图谱及聚类分析研究,以期为喜树叶的质量控制提供依据。
1 材料
1.1 仪器
e2695型高效液相色谱(HPLC)仪,包括2998 DAD检测器、Empower色谱数据工作站(美国Waters公司);CPA225D型十万分之一电子分析天平(北京赛多利斯仪器有限公司);AY220型万分之一电子分析天平(日本Shimadzu公司);DK-98-Ⅱ型电热恒温水浴锅(天津市泰斯特仪器有限公司)。
1.2 试剂
槲皮素对照品(批号:100081-201509,纯度:98.6%)、山柰素对照品(批号:110861-201310,纯度:93.2%)均购自中国食品药品检定研究院;乙腈为色谱纯,其余试剂均为分析纯,水为纯化水。
1.3 药材
喜树叶药材样品均采于贵州省,共14批(见表1),经贵州省食品药品检验所李阳教授鉴定为真品。
2 方法与结果
2.1 色谱条件
色谱柱:Gemini-NX C18(250 mm×4.6 mm,5 μm);流动相:乙腈(A)-0.2%磷酸溶液(B),梯度洗脱(0~20 min,22%A;20~40 min,22%→35%A;40~60 min,35%A);流速:1.0 mL/min;检测波长:370 nm;柱温:30℃;进样量:10 μL。
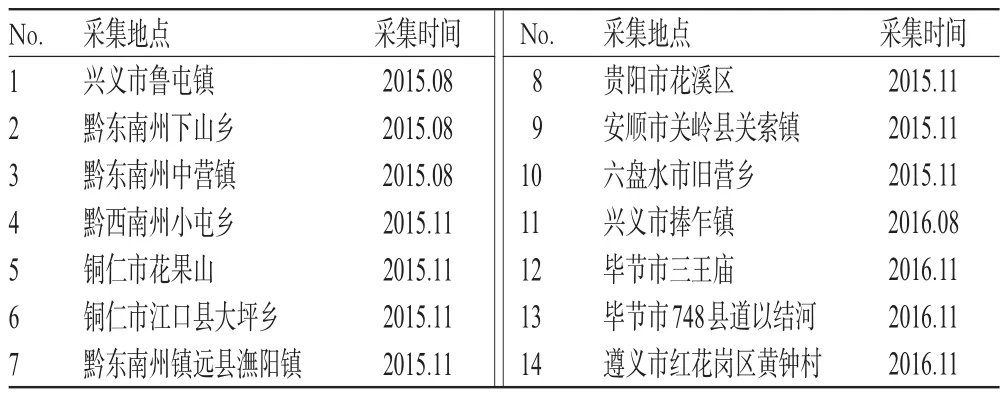
表1 喜树叶药材来源Tab 1 Source of C.acuminate
2.2 溶液的制备
2.2.1 混合对照品溶液 精密称取槲皮素对照品14.01 mg,加甲醇定容至100 mL,量取5 mL,置于25 mL量瓶中,制成槲皮素对照品溶液;精密称取山柰素对照品适量,置于上述装有槲皮素对照品溶液的25 mL量瓶中,摇匀,制成槲皮素、山柰素质量浓度分别为27.63、19.96 μg/mL的混合对照品溶液。
2.2.2 供试品溶液 将药材样品置于干燥的室内自然风干,粉碎后过4号筛,备用。取药材样品粉末约0.5 g,精密称定,置于具塞锥形瓶中,精密加入5%盐酸甲醇溶液50 mL,称定质量,水浴回流1 h,取出,放冷;再次称定质量,用5%盐酸甲醇溶液补足减失的质量,摇匀,滤过,取续滤液,经0.45 μm微孔滤膜滤过,取续滤液,即得。
2.3 方法学考察
2.3.1 精密度试验 取“2.2.1”项下混合对照品溶液适量,按“2.1”项下色谱条件连续进样测定6次,以山柰素的保留时间和峰面积为参照,记录各共有峰相对保留时间和相对峰面积。结果,10个共有峰相对保留时间的RSD<0.3%,相对峰面积的RSD<1.2%(n=6),表明仪器精密度良好。
2.3.2 稳定性试验 取“2.2.2”项下供试品溶液(No.5)适量,分别于室温下放置0、2、4、8、12、24 h时进样测定,以山柰素的保留时间和峰面积为参照,记录各共有峰相对保留时间和相对峰面积。结果,10个共有峰相对保留时间的RSD<0.4%,相对峰面积的RSD<1.1%(n=6),表明供试品溶液室温放置24 h内基本稳定。
2.3.3 重复性试验 精密称取同一批样品(No.5)适量,按“2.2.2”项下方法制备供试品溶液,共6份,再按“2.1”项下色谱条件进样测定,以山柰素的保留时间和峰面积为参照,记录各共有峰相对保留时间和相对峰面积。结果,10个共有峰相对保留时间的RSD<0.7%,相对峰面积的RSD<0.6%(n=6),表明本方法重复性良好。
2.4 HPLC指纹图谱的生成与相关数据分析
2.4.1 HPLC指纹图谱的生成 取14批药材样品各适量,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,采用《中药色谱指纹图谱相似度评价系统》(2004 A)对14批药材样品的HPLC图谱进行分析,得HPLC指纹图谱,详见图1、图2。

图1 14批药材样品HPLC叠加指纹图谱Fig 1 HPLC superposed fingerprints of 14 batches of samples

图2 药材样品HPLC对照指纹图谱Fig 2 HPLC control fingerprint of samples
2.4.2 相似度与各共有峰相关数据分析 采用《中药色谱指纹图谱相似度评价系统》(2004 A)对14批药材样品的HPLC图谱进行比较分析。14批药材样品相似度评价结果见表2;各共有峰的相对保留时间和相对峰面积见表3、表4。
2.5 聚类分析
采用SPSS 18.0统计软件与欧氏距离分层聚类法中的Ward法对14批药材样品指纹图谱共有峰相对峰面积进行聚类分析[4],详见图3。结果,14批药材样品分为3大类,其中3、4、6、7、9、11、12、13批药材样品为一类,8、10批药材样品为一类,1、2、5、14批药材样品为一类,但1和2、5、14批药材样品欧氏距离稍大,也可以将1批药材样品再细分为一类。在指纹图谱相似度评价中,1批药材样品与对照图谱的相似度均<0.90;其余13批药材样品与对照图谱的相似度>0.90。由此可知,聚类分析结果和相似度评价结果基本一致,两种方法相互印证。
3 讨论
3.1 样品提取条件的优化
笔者以61%乙醇溶液超声处理30 min制成供试品溶液,得到的色谱图峰较少。后改以5%盐酸甲醇溶液水浴回流1 h提取得到的色谱图中色谱峰较多,且分离度均较好,因此本试验以上述方法进行样品提取。
3.2 色谱条件的优化
笔者参考文献[5],分别以甲醇-0.4%磷酸溶液(50∶50,V/V)和乙腈-0.3%磷酸溶液(24∶76,V/V)为流动相,进行等度洗脱。结果,色谱峰部分重叠,且特征峰少,经反复试验,最终确定以乙腈-0.2%磷酸溶液作为流动相,进行梯度洗脱,药材样品所得特征峰多,且峰形较好。
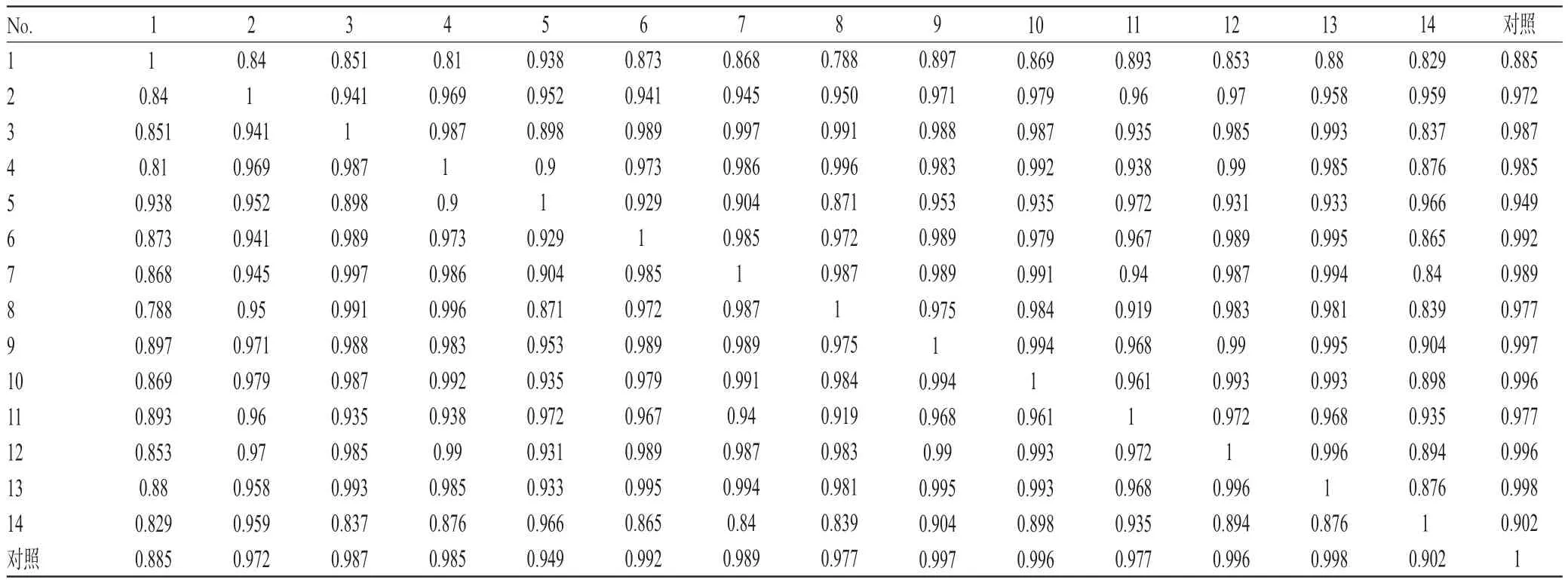
表2 14批药材样品相似度评价结果Tab 2 Similarity evaluation of 14 batches of samples

表3 14批药材样品HPLC图谱共有峰的相对保留时间Tab 3 The relative retention time for common peaks in HPLC fingerprints of 14 batches of samples

表4 14批药材样品HPLC图谱共有峰的的相对峰面积Tab 4 The relative peak areas for common peaks in HPLC fingerprints of 14 batches of samples
3.3 测定波长的选择
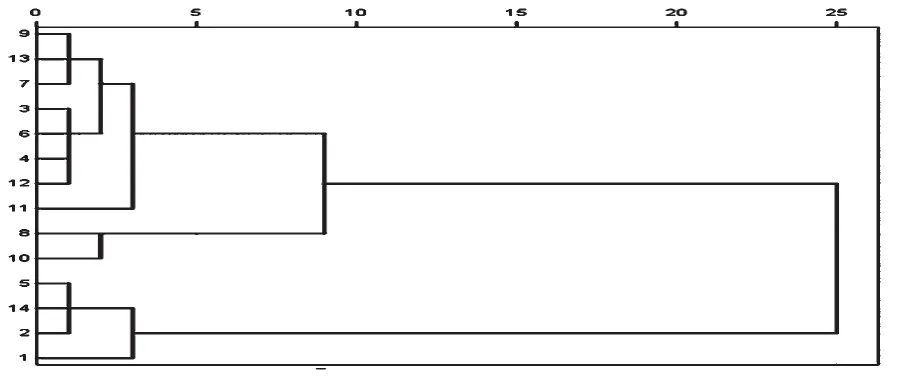
图3 14批药材样品聚类分析树状图Fig 3 The cluster analysis dendrogram for 14 batches of samples
试验中,笔者应用二极管阵列检测器对药材样品进行了全波段检测,结果在370 nm波长处,相对吸收峰比较多,基本能反映出药材样品的整体效果,在线性梯度洗脱时,基线基本平稳,所以确定检测波长为370 nm。
笔者通过建立喜树叶药材的指纹图谱,采用相似度评价软件对来自贵州不同产地的喜树叶药材进行比较分析,可知贵州不同产地14批喜树叶药材指纹图谱总体差异较小,但可能由于喜树叶采集的季节不同,使得个别样品(No.1)的指纹图谱与对照图谱相似度较低,质量较为一般。本研究所建鉴别方法可为完善喜树叶质量标准提供借鉴,进一步为喜树叶药材收入《贵州省中药材、民族药材质量标准》提供参考。
[1] 中国科学院中国植物志编辑委员会.中国植物志[M].北京:科学出版社,1983:144.
[2] 国家中医药管理局《中华本草》编辑委员会.中华本草:第5册[M].上海:上海科学技术出版社,1999:733.
[3] 阎秀峰,王洋,于涛,等.喜树叶中喜树碱含量的高效液相色谱分析[J].分析测试学报,2002,21(2):15-18.
[4] 黄玮,周云庆,杨洪元,等.胆黄润肠丸指纹图谱的聚类分析[J].中国医院药学杂志,2016,36(9):719-723.
[5] 秦庆芳,粟本超.广西不同地区喜树嫩叶中喜树碱含量测定[J].医药导报,2015,34(3):388-391.
Study on HPLC Fingerprint and Cluster Analysis of the Leaves of Camptotheca acuminate in Guizhou
LIU Zhihai,FAN Hongmei,ZOU Lan,LONG Lin,JIN Zhao,YU Lan(Guizhou Qianxinan Prefecture Center for Food and Drug Inspection,Guizhou Xingyi 562400,China)
OBJECTIVE:To establish HPLC fingerprint for the leaves of Camptotheca acuminante in Guizhou.METHODS:HPLC method was performed.The determination was performed on Gemini-NX C18column with mobile phase consisted of acetonitrile-0.2%phosphoric acid(gradient elution)at the flow rate of 1.0 mL/min.The detection wavelength was set at 370 nm,and the column temperature maintained at 30℃.The sample size was 10 μL.Using sorbitol as a reference,HPLC fingerprints of 14 batches of the leaves of C.acuminante were determined.The chromatographic fingerprint was analyzed with Similarity Evaluation System for Chromatographic Fingerprint of TCM(2004 A)in terms of common peak indentification,similarity evaluation and cluster analysis.RESULTS:There were 10 common peaks in HPLC fingerprints for 14 batches of the leaves of C.acuminate.And the similarity of 13 batches of the leaves of C.acuminate was greater than 0.90,and that of another one was less than 0.90.The leaves of C. acuminate were classified into 3 groups.CONCLUSIONS:The established fingerprint can provide reference for identification and quality evaluation of the leaves of C.acuminate.
The leaves of Camptotheca acuminante;Fingerprint;Cluster analysis;HPLC
R917
A
1001-0408(2017)24-3412-03
2017-01-18
2017-04-26)
(编辑:张 静)
贵州省中药材、民族药材质量标准研究课题(No. DBXZYC52032)
*主管中药师。研究方向:中药、中成药质量标准。E-mail:19889504@qq.com
DOI10.6039/j.issn.1001-0408.2017.24.29
猜你喜欢
小哥白尼(趣味科学)(2021年11期)2021-02-28
小天使·一年级语数英综合(2020年10期)2020-12-16
北方药学(2020年12期)2020-04-30
中华肺部疾病杂志(电子版)(2020年1期)2020-01-07
中成药(2019年12期)2020-01-04
中成药(2019年12期)2020-01-04
中国药理学与毒理学杂志(2019年4期)2019-08-12
中成药(2017年9期)2017-12-19
中成药(2017年7期)2017-11-22
中成药(2017年4期)2017-05-17
