
Transcriptome analysis of Verticilliumlecanii based on RNA-Seq technology
2017-08-29 11:00ZHANGMouGAOXiHAOKaiqiangTANQingPANGRenyiLANMingxianLIZhaoboXIATaoLUWufengLIJianyiWUGuoxing
生物安全学报 2017年3期
ZHANG Mou, GAO Xi, HAO Kaiqiang, TAN Qing, PANG Renyi, LAN Mingxian, LI Zhaobo, XIA Tao, LU Wufeng, LI Jianyi, WU Guoxing
College of Plant Protection, Yunnan Agricultural University, Kunming, Yunnan 650201, China
bor L. LÖVEI, 郭莹)
Transcriptome analysis ofVerticilliumlecaniibased on RNA-Seq technology
ZHANG Mou, GAO Xi*, HAO Kaiqiang, TAN Qing, PANG Renyi, LAN Mingxian, LI Zhaobo, XIA Tao, LU Wufeng, LI Jianyi, WU Guoxing*
CollegeofPlantProtection,YunnanAgriculturalUniversity,Kunming,Yunnan650201,China
【Aim】Verticilliumlecaniiis a well-known biocontrol agent of fungal phytopathogens as well as insect pests. It is important to investigate the molecular basis of the pathogenicity. 【Method】 We analyzed the transcriptome ofV.lecaniiisolated fromBoettcheriscaperegrina. 【Result】 A total of 1634670 high-quality reads were produced by RNA-seq method with Q20 percentage of 89.47% and the GC percentage of 48.50%. The Nr, Swiss-Prot, Gene Ontology (GO), Clusters of Orthologous Group (COG), and Kyoto Encyclopedia of Genesand Genomes (KEGG) databases were used to annotate unigenes functions. Of the 14856 Unigenes assembled, 1467, 9379, 6518, and 4188 were mapped by Nr, Swiss-Prot, COG, and KEGG, respectively. In the GO database, 21403 Unigenes were classified into 38 functional groups, belonging to three main categories: cellular components (1369), molecular functions (1679), and biological processes (1928). Based on COG annotation, 6518 Unigenes were classified into 24 functional categories. The important functional groups identified via GO and COG enrichment were responsible for general function prediction only, secondary metabolites biosynthesis, transport and catabolism, and signal transduction mechanisms. Unigenes mapped to 108 KEGG pathways, most of which were associated with metabolic pathways and biosynthesis of secondary metabolites. 【Conclusion】 Our results will be helpful to find the pathogenic factors ofV.lecaniiand enrich its pathogenic mechanism.
Verticilliumlecanii; RNA-Seq; transcriptome
Verticilliumlecaniiis a genus of fungi in the division Ascomycota, and is an anamorphic form of the family Plectosphaerellaceae.V.lecaniiis a well-known entomopathogenic fungus with wide infection range including homopteran insects and other arthropod groups. The entomopathogenic fungi must penetrate the insect cuticle, which comprises mainly chitin fibrils embedded in a protein matrix. The insect infection occurs by a combination of mechanical pressure, via appressorium formation, and enzymatic degradation.V.lecaniias a biological control agents (Lietal.,2004) are suggested as replacement to pesticides, which are known to have negative effects on the environment and non-target organisms (Harmanetal.,1993).V.lecaniishows potential biological control agent against numerous pest insect orders, such as Coleoptera, Orthoptera, Homoptera, and Lepidoptera. Interestingly,V.lecaniiinduced host defense reactions without showing the pathogenicity phenotype (Benhamou & Brodeur,2000).Boettcheriscaperegrina(Diptera: Sarcophagidae) is regarded as a medium to spread pathogens. Flies, one of the largest biomass in the world, could be found nearly in every corner of the earth. It is difficult to take effective measures for controlB.peregrina(Goffetal.,1991). The extensive use of the fast and effective pesticides had been forbidden due to death of non-target species, accumulation of pesticide residues in the environment and food, and build-up of pesticide resistance in the target species. Biological control agents are developing quickly, such as different attractants or different entomopathogenic fungi likeBeauveriabassianaandV.lecanii.V.lecaniican kill host by production of active compounds like chitinases, proteins and polysaccharide degrading enzymes (amylase, protease, and lipase) (Hasanetal.,2013; Xuetal.,2011; Yuetal.,2015; Zhuetal.,2008). However, the roles that the active compounds could play in the infection process are still not fully understood. Furthermore, the genetic and molecular basis of the interaction of entomopathogenic fungus, includingV.lecaniiand the insect hosts are not fully understood.
In order to better understand the mechanism ofV.lecaniientomopathogenicity, we studied the transcriptome ofV.lecaniito reveal the pathogenic mechanism ofV.lecaniiinB.peregrina. Our aim is to identify target genes for genetic manipulation to develop more efficientV.lecaniistrains for specific insect pests control.
1 Materials and Methods
1.1 Culturing ofV.lecanii
V.lecaniistrain KMZW-1, isolated fromB.peregrinafrom the collection of the Yunnan Agricultural University, was used in our experiments. Cultures were initially grown on potato dextrose agar (PDA) (Tanetal.,2015) for 7 days at 29 ℃. To obtain conidia, the cultured isolate was incubated in 100 mL Czapek liquid medium in 250 mL incubation bottles for 5 days at 29 ℃. The spore suspension obtained was diluted to approximately 1×107/mL (Yuetal.,2015) with sterile distilled water prior to inoculation.
1.2 Extraction and purification of total RNA
Total RNA was extracted in TRIzol reagent (GIBCO BRL) according to the manufacturer′s instructions. The total RNA was dissolved in 50 μL RNase free water. RNA quality was checked on agarose gels (1.5%) and concentration and OD260/280 value was determined Nano-drop (Thermo, NANODROP 2000). The total RNA was stored at -80 ℃ until further use.
1.3 Library preparation for RNA-Seq
Libraries were prepared using RNA-Seq sample preparation kit from Illumina and poly (A) mRNA was enriched from total RNA using oligo (dT) beads. Fragmentation buffer was added for breaking mRNA to short fragments. Using these fragments as template, first-strand cDNA was synthesized by reverse transcription with oligo (dT) random hexamer primer. The second-strand was synthesized using buffer, dNTPs, RNase H and DNA polymerase I according to kit manufacturer instructions. Short fragments were purified with QiaQuick PCR extraction kit (Qiagen, USA) and resolved with ddH2O. Sequencing adaptors were added and amplified with PCR. Agarose gel electrophoresis was used to select the fragments collected as sequencing templates. Finally, the cDNA library was constructed and sequenced on the Illumina HiSeqTM 2000 platform (GENE DENOVO, Guangzhou).
1.4 Quality control and splicing of RNA sequence
RNA-Seq reads were subjected to quality filtering using base calling on the basis of the criterion of a minimum read length of 30, and good-quality reads were obtained with Q20 (sequencing error rate lower than 1%) and discarding sequences of adapters reads sequence and reads with bases ‘N’ more than 10% for all reads. Before read alignment, rRNA fragments were filtered from the transcriptomics data by the use of base calling software and the default rRNA database included in the software package. Reads were aligned to the genome ofV.lecanii, and gene annotations were obtained from the four protein database of bacterial genomes (Nr, Swiss-Prot, KEGG and COG) by BLAST.
1.5 Gene ontology and functional annotation
After clustering and assembly, to identify similarities between the sequences deposited in public databases, a BLAST (basic local alignment aearch tool) (Conesaetal.,2005) alignment was performed by searching the Non-redundant (Nr), Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG), and Clusters of Orthologous Groups (COG) protein databases with a cut offE-value≤1e-5. The Unigene sequences were also translated into proteins via Trinity software (Grabherretal.,2011). Trinity software was also used with anE-value≤1e-5, to annotate the Unigene′major Gene Ontology (GO) categories, including molecular functions, biological processes, and cellular components. However, when the Trinity software cannot compare by the four databases (Nr, Swiss-Prot, KEGG and COG), the ETScan (Iselietal.,1999) software was used to confirm with the direction of protein. Gene expression levels were determined by Fragments per Kilobase of gene per Million mapped fragments (FPKM). Unigene expression levels were determined by using RPKM; RPKM= (1000000×C)/(N×L/1000), (RPKM was represented expression level of Unigene gene,Cwas reads number of Unigene gene,Nwas mapped in total reads number of Unigene gene,Lwas represented total bases of Unigene gene).
2 Results
2.1 Sequence assembly
After stringent quality checks and tag cleaning of the total of 42692122 reads, 1634670 high-quality sequences were generated. Any short, low-quality sequences and adapters reads sequence and reads with bases ‘N’ more than 10% were removed, total nucleotides were 5336515250 nt, Q20 and ‘G+C’ content of 89.47%, 48.50% respectively. 21403 Unigenes were derived from cluster assembly, with an average length of 1296.08 bp, average ‘G+C’ content of 52.09%, the value-N50 is 2143 and total assembled bases 27739917. Over 1000 reads were mapped in 704 unigenes with the average coverage of 97.23% (Fig.1).
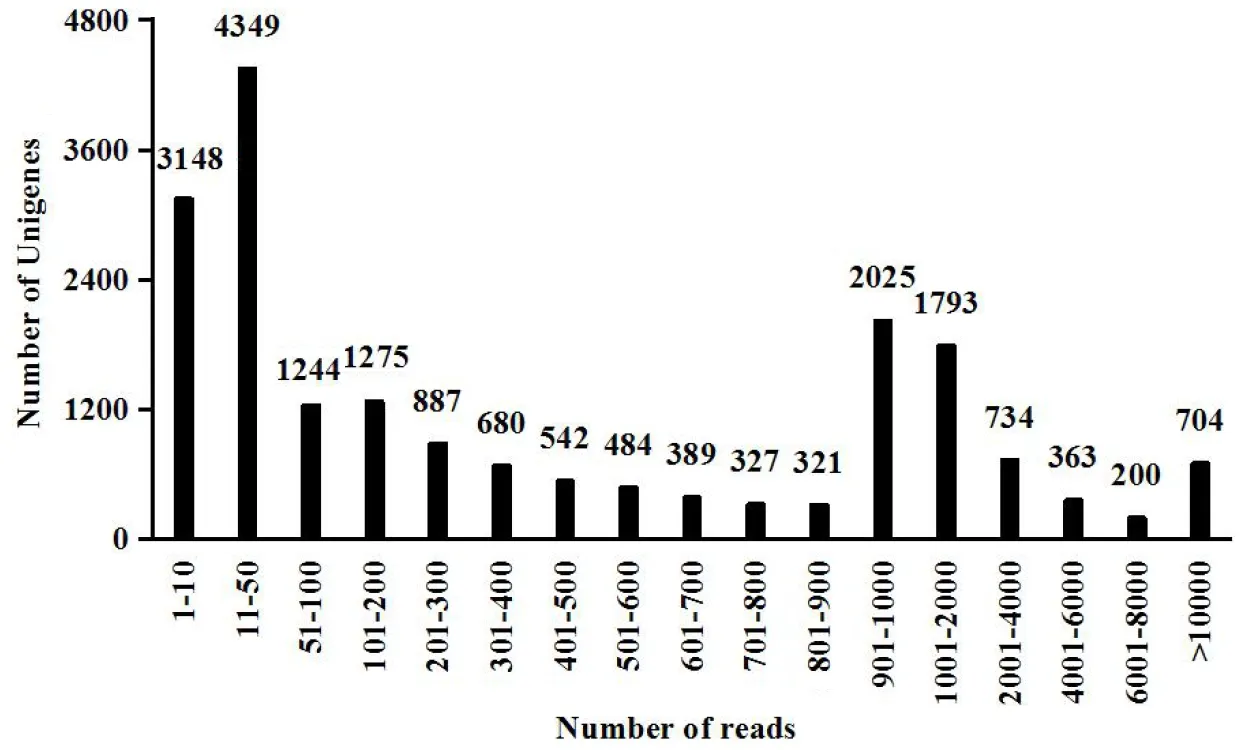
Fig.1 Mapped reads in Unigene
2.2 Functional annotation and classification
A sequence similarity search was conducted against the Nr, Swiss-Prot, KEGG and COG databases, usingBlast2GOthat specifiedE-values of less than 10-5. A total of 14856 Unigenes (69.4%) had annotation information in one or more of the Nr, Swiss-Prot, GO, KEGG, and COG databases. The analysis showed that 14679 Unigenes had significant matches in the Nr database; 9379 Unigenes had significant matches in the Swiss-Prot database; 6518 Unigenes had significant matches in the KEGG database; 4188 Unigenes had significant matches in the COG database. A total of 3048 Unigenes could be matched in COG and KEGG, 6493 Unigenes could be matched in COG and Nr, 6200 Unigenes could be matched in COG and Swiss-Prot, 4187 Unigenes could be matched in KEGG and Nr, 3865 Unigenes could be matched in KEGG and Swiss-Prot, 6181 Unigenes could be matched in Nr and Swiss-Prot, and 3002 Unigenes could be mapped in all four databases (Fig.2). However, 6547 Unigenes had not matches in any of these databases. Those without database hits were likely to include non-coding RNAs, genes whose sequences did not capture regions that contained conserved functional domains, or protein coding genes that were novel in the database. Mapping revealed that 21398 predicted genes were expressed. Approximately 71.63% (15328) of genes were expressed with FPKM>1.
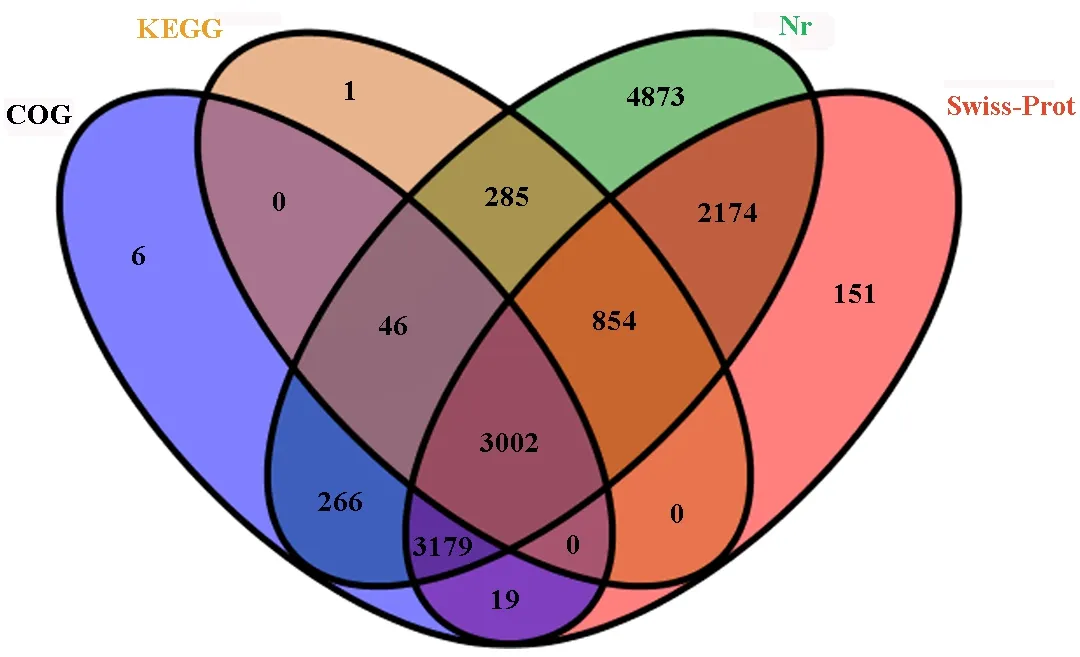
Fig.2 Venn diagram of overlaps for Unigenes annotation in four protein databases
The identified functional classes of the Unigenes were subjected to GO enrichment analysis. According to the results of sequence alignments, 21403 differential Unigenes were classified into 38 functional groups, belonging to three main categories (Fig.3): cellular components (1369), molecular functions (1679), and biological processes (1928). In the cellular component category, 784 Unigenes were localized to cell and cell part, 200 to organelle and 89 to macromolecular complex. In the molecular function category, a large number of Unigenes were involved in catalytic activity and binding. In the biological process, a large number of Unigenes were involved in metabolic process and cellular process. In the biological processes category, most Unigenes were involved in metabolic process and cellular process. Several Unigenes were found to participate in locomotion, immune system process, death, and growth. In summary, these results showed that most of the identified Unigenes were responsible for catalytic activity associated with biological regulation and metabolism.
The biological functions of Unigenes were performed to categorize in KEGG pathway enrichment analysis. A total of 4188 Unigenes were sited to 108 KEGG pathways. The metabolic pathways contained the highest Unigenes numbers (1222, 29.18%), the 498 Unigenes (11.89%) involved in the biosynthesis of secondary metabolites, 166 Unigenes (3.96%) were connected to ribosome, the 127 Unigenes (3.03%) were part of purine metabolism, the 120 Unigenes (2.87%) were oxidative phosphorylation, the 107 Unigenes (2.55%) were spliceosome and the least number of Unigenes (2, 0.05%) were connected to C5-branched dibasic acid metabolism.These results suggested that metabolic pathways, biosynthesis of secondary metabolites, ribosome, and spliceosome should be associated with theB.peregrinasusceptible response toV.lecanii.
Unigenes were classified into different COG classes, according to COG database. 6518 Unigenes could be classified into 24 functional categories based on COG annotation. The Unigenes of 24 functional categories were sequenced based on their number, primarily included general function prediction only (2100), amino acid transport and metabolism (1079), carbohydrate transport and metabolism (942), secondary metabolites biosynthesis, transport and catabolism (775), inorganic ion transport and metabolism (682) (Fig.4). The Unigenes involved in signal transduction mechanisms, secondary metabolites biosynthesis, transport and catabolism, and defense mechanisms are interUnigenes because Unigenes in these functional categories might participate in secondary metabolites and complex signaling pathways in response to the pathogen.
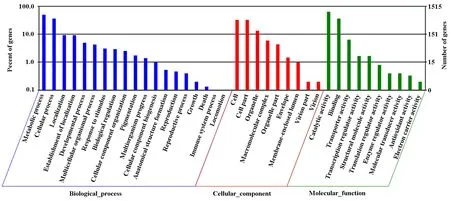
Fig.3 The gene ontology (GO) categories of Unigene
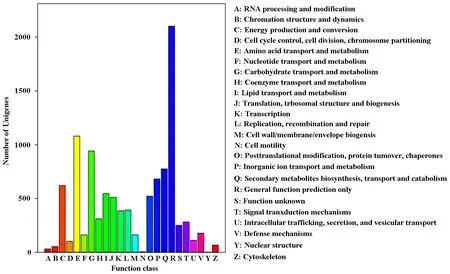
Fig.4 Classifications for clusters of orthologous groups (COGs)
2.3 Selection of the core candidates related to chitin
The chitin is an important composition in the shells of insects, and the chitinase is a possbile key to pathogenic factor of insecta pathogenic fungus. The chitinase expression ofV.lecaniiwas higher than other enzymes (Hall,1981; Verhaaretal.,1996), for instance,Beauveriabassiana,Metarhiziumanisopliae. We selected several chitin pathways related Unigenes involved in three KEGG pathways (metabolic pathways, biosynthesis of secondary metabolites and ribosome), followed by selection of COG, GO and Nr annotation of the core candidates related toB.peregrinasusceptibility toV.lecanii. We obtained 6 Unigenes (Table 1) about the genes of chitinase.
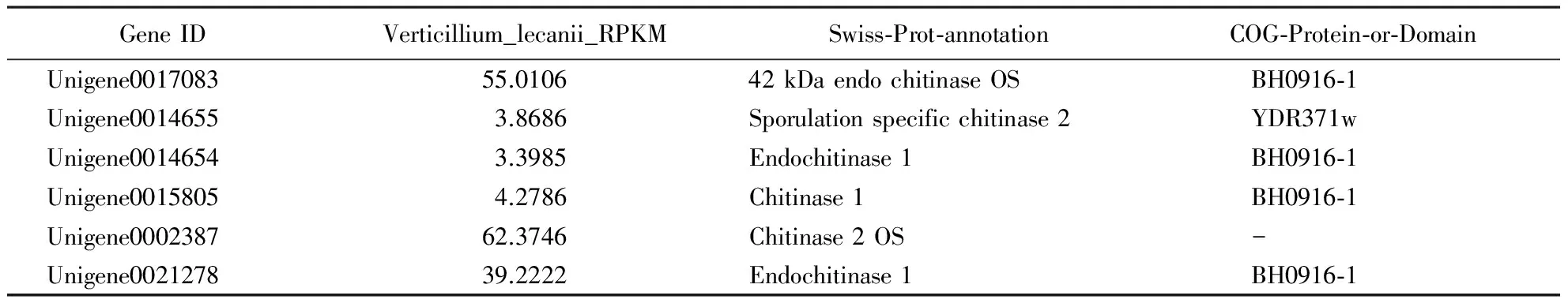
Table 1 The candidate Unigenes related to chitinase
2.4 Identification of Unigenes-SSRs
We identified 1476 SSRs in 21403 (6.9%) Unigenes. Tri-nucleotide was the highest SSRs (835), followed by di-nucleotide (308), tetra-nucleotide (243), penta-nucleotide (58) and hexa-nucleotide (32). Molecular marker is important resources for constructing genetics maps. Special Unigenes SSRs have high efficiency and a low cost, and can improve effectiveness of the work for us. The distribution of the different SSR repeat types as a proportion of the total SSRs was represented in Fig.5.
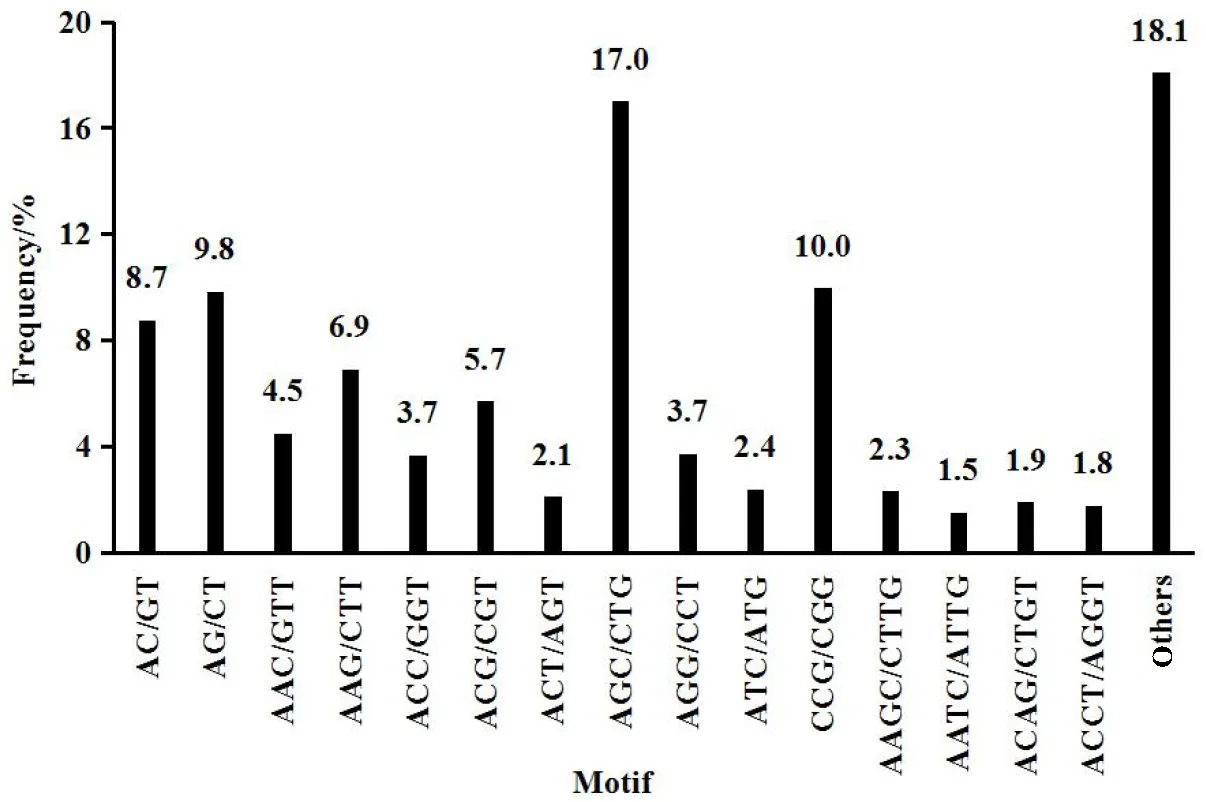
Fig.5 The distribute of SSRs
3 Discussion
Entomopathogenic fungi are natural biological control agents for many insects and other arthropods. By spreading diseases,B.peregrinais one of most sanitation pest in cities.V.lecaniiis an entomopathogenic fungus with a various hosts insects. In order to widen our knowledge of the pathogenecity mechanism ofV.lecaniiagainstB.pereqrinatranscriptome analysis ofV.lecaniiisolated fromB.pereqrinawas performed.
The insect cuticle major components are chitin and cuticle protein (Moussian,2010). Cuticle protein is an important to the development of insect and invertebrates, contributes much to the stress resistance, drug resistance, and immunity of insects. The genes coding for cuticle proteins were induced to resist infection against entomopathogenic fungi in adverse environment conditions (Puracetal.,2008; Zhangetal.,2008). Chitinases were widely distributed in plants, bacteria, fungi, insects and vertebrates (Luetal.,2005). Chitinase in collaboration with proteases were associated with different stages of the life cycle of entomopathogenic fungi, such as germination, hyphal growth, morphogenesis, and defence against competitors (Adams,2004). Chitinases mainly responsible physiological functions as degradation the fungal cellular walls or the exoskeletons of arthropods used as nutrient sources to development; rebuilding of cell walls during hyphae growth, branching, hyphae fusion, autolysis and competence (Adams,2004; Yangetal.,2007). Chitinases had critical functions in the process of growth and degradation of the fungal cell wall and insect cuticle, with chitin being a major component of both. Fungal virulence was often determined with the extracellular chitinases (Yuetal.,2015). The germination of the spore of entomopathogenic fungi on the insect cuticle followed by penetration and vigorous proliferation of fungal hyphae ultimately desiccate the host insects. The life cycle of the fungi continues with the production of infective spores that can insert themselves immediately into the insect cuticle to repeat the cycle, after a period of dormancy.
In transcriptome profiling ofGossypiumbarbadenseinoculated withVerticilliumdahliaethe overwhelming majority of the Unigenes were mapped in the biological process (Zhangetal.,2013), and many of these were related to metabolic processes, cellular processes, localization, establishment of localization, developmental process, multicellular organismal process, responses to stimuli and biological regulation, metabolic and cellular processes and responses to stimuli. The reported results suggested that similar genes could be expressed in both susceptible and resistant insects in responses toV.lecanii(Zhangetal.,2013).
We reported here the transcriptomic profiling theV.lecaniistrain isolated fromB.pereqrina. There were some resistant or defense response related genes (purine metabolism, oxidative phosphosphorylation) in our data. These results suggested that there migh be an overlap among the susceptible responses of host insects and fungal pathogens. In addition, functional COG categories showed that the identified Unigenes were mainly involved in function prediction only, namely amino acid transport and metabolism, carbohydrate transport and metabolism, secondary metabolites biosynthesis, transport and catabolism, inorganic ion transport and metabolism, similar to the results obtained in the biological process enrichment. For further understand the biological functions of the identified Unigenes, KEGG pathway enrichment analysis was carried out. Hundred and eight pathways were examined. The two pathways with the most related Unigenes were metabolic pathways and biosynthesis of secondary metabolites pathways (a total of 1720, 41.07%). It was reported that the phenylalanine metabolism pathway played a critical role in defense response toV.dahlia(Gayosoetal.,2010). In our experiment the phenylalanine metabolism pathway showed a lot related Unigenes expression (54, 1.29%), and from this result we infer the phenylalanine metabolism pathway was an important part to infect the host ofV.lecanii. The overwhelming majority of the Unigenes (1222, 29.18%) were mapped in metabolic pathway versus resistance responses (2329) of the hosts againstV.lecanii.
The cuticle is the first barrier for insects to defend against pathogen infection, in addition to being indispensable for maintaining the shape and mobility of insects (Delon & Payre,2004; Zhuetal.,2008). During the penetration process, entomopathogenic fungi can produce several chitinases, some of which are important chitinous degrading enzymes and act synergistically with proteases to hydrolyze insect cuticle. Chitinases component of the cell walls of fungi and insects, furthermore they are a key pathogenicity determinants of entomopathogeic fungi (Rocha-Pinoetal.,2011). Chitinous receptors (OsCEBiPandAtCERK1) had been identified in bacterial, fungi, virus, and plants (Liuetal.,2012). Chitinases are not only playing important physiological and ecological roles in defense mechanisms in response to pathogens and abiotic stress, but also involved in nutrition and pathogenesis. Fungal pathogenesis is a complex process that includes adherence, penetration, and digestion and requires the production of hydrolytic enzymes that penetrate the chitinous cell wall or cuticle. The unigenes of chitinesaes (Table 1) were suggested as the core candidates for further experiments to improveV.lecaniias a biological control agent.
ADAMS D J, 2004. Fungal cell wall chitinases and glucanases.Microbiology, 150: 2029-2035.
BENHAMOU N, BRODEUR J, 2000. Evidence for antibiosis and induced host defense reactions in the interaction betweenVerticilliumlecaniiandPenicilliumdigitatum, the causal agent of green mold.Phytopathology, 90(9): 932-943.
CONESA A, GOTZ S, GARCIA-GOMEZ J M, TEROL J, TALON M, ROBLES M, 2005. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research.Bioinformatics, 21(18): 3674-3676.
DELON I, PAYRE F, 2004. Evolution of larval morphology in flies: get in shape with shavenbaby.TrendsinGenetics, 20: 305-313.
GAYOSO C, POMAR F, NOVO-UZAL E, MERINO F, DE ILARDUYA O M, 2010. The Ve-mediated resistance response of the tomato toVerticilliumlecaniiinvolves H2O2, peroxidase and lignins and drives PAL gene expression.BMCPlantBiology, 10: 232.
GOFF M L, BROWN W A, HEWADIKARAM K A, OMORI A I, 1991. Effect of heroin in decomposing tissues on the development rate of boettcherisca-peregrina (Diptera, sarcophagidae) and implications of this effect on estimation of postmortem intervals using arthropod development patterns.JournalofForensicSciences, 36(2): 537-542.
GRABHERR M G, HAAS B J, YASSOUR M, LEVIN J Z, THOMPSON D A, AMIT I, ADICONIS X, FAN L, RAYCHOWDHURY R, ZENG Q D, CHEN Z H, MAUCELI E, HACOHEN N, GNIRKE A, RHIND N, DI PALMA F, BIRREN B W, NUSBAUM C, LINDBLAD-TOH K, FRIEDMAN N, REGEV A, 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome.NatureBiotechnology, 29(7): 644-652.
HALL R A, 1981. The fungusVerticilliumlecaniias a microbial insecticide against aphids and scales∥BURGES H D.Microbialcontrolofpestsandplantdieases1970-1980. London: Academic Press: 483-498.
HARMAN G E, HAYES C K, LORITO M, BROADWAY R M, DIPIETRO A, PETERBAUER C, TRANSMO A, 1993. Chitinolytic enzymes of trichoderma-harzianum — purification of chitobiosidase and endochitinase.Phytopathology, 83(11): 313.
HASAN S, AHMAD A, PURWAR A, KHAN N, KUNDAN R, GUPTA G, 2013. Production of extracellular enzymes in the entomopathogenic fungusVerticilliumlecanii.Bioinformation, 9(5): 238-242.
ISELI C, JONGENEEL C V, BUCHER P, 1999. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences.ProceedingsoftheSeventhISMB, 99(2): 138-148.
LI D C, ZHANG S H, LIU K Q, LU J, 2004. Purification and partial characterization of a chitinase from the mycoparasitic fungusTrichotheciumroseum.JournalofGeneralandAppliedMicrobiology, 50(1): 35-39.
LIU T T, LIU Z X, SONG C J, HU Y F, HAN Z F, SHE J, FAN F F, WANG J W, JIN C W, CHANG J B, ZHOU J M, CHAI J J, 2012. Chitin-induced dimerization activates a plant immune receptor.Science, 336: 1160-1164.
LU Z X, LAROCHE A, HUANG H C, 2005. Isolation and characterization of chitinases fromVerticilliumlecanii.CanadianJournalofMicrobiology, 51: 1045-1055.
MOUSSIAN B, 2010. Recent advances in understanding mechanisms of insect cuticle differentiation.InsectBiochemistryandMolecularBiology, 40: 363-375.
PURAC J, BURNS G, THORNE M A, GRUBOR-LAJSIC G, WORLAND M R, CLARK M S, 2008. Cold hardening processes in the Antarctic springtail,Cryptopygusantarcticus: clues from a microarray.JournalofInsectPhysiology, 54: 1356-1362.
ROCHA-PINO Z, VIGUERAS G, SHIRAI K, 2011. Production and activities of chitinases and hydrophobins fromLecanicilliumlecanii.BioprocessandBiosystemsEngineering, 34(6): 681-686.
TAN G, LIU K, KANG J, XU K, ZHANG Y, HU L, ZHANG J, LI C, 2015. Transcriptome analysis of the compatible interaction of tomato withVerticilliumdahliaeusing RNA-sequencing.FrontiersinPlantScience, 6: 428.
VERHAAR M A, HIJWEGEN T, ZADOKS J C, 1996. Glasshouse experiments on biocontrol of cucumber powdery mildew (Sphaerothecafuliginea) by the mycoparasitesVerticilliumlecaniiandSporothrixrugulosa.BiologicalControl, 6(3): 353-360.
XU X, YU Y, SHI Y, 2011. Evaluation of inert and organic carriers forVerticilliumlecaniispore production in solid-state fermentation.BiotechnologyLetters, 33(4): 763-768.
YANG J, TIAN B, LIANG L, ZHANG K Q, 2007. Extracellular enzymes and the pathogenesis of nematophagous fungi.AppliedMicrobiologyandBiotechnology, 75: 21-31.
YU G, XIE L Q, LI J T, SUN X H, ZHANG H, DU Q, LI Q Y, ZHANG S H, PAN H Y, 2015. Isolation, partial characterization, and cloning of an extracellular chitinase from the entomopathogenic fungusVerticilliumlecanii.GeneticsandMolecularResearch, 14(1): 2275-2289.
ZHANG J, GOYER C, PELLETIER Y, 2008. Environmental stresses induce the expression of putative glycine-rich insect cuticular protein genes in adultLeptinotarsadecemlineata.InsectMolecularBiology, 17: 209-216.
ZHANG Y, WANG X F, DING Z G, MA Q, ZHANG G R, ZHANG S L, LI Z K, WU L Q, ZHANG G Y, MA Z Y, 2013. Transcriptome profiling ofGossypiumbarbadenseinoculated withVerticilliumdahliaeprovides a resource for cotton improvement.BMCGenomics, 14: 637.
ZHU Y, PAN J, QIU J, GUAN X, 2008. Isolation and characterization of a chitinase gene from entomopathogenic fungusVerticilliumlecanii.BrazilianJournalofMicrobiology, 39(2): 314-320.
2016-08-08 接受日期(Accepted): 2017-05-11
国家自然科学基金项目(31160379)
张某, 男, 硕士研究生。 研究方向: 有害生物治理。 E-mail: zhangmengtao01@outlook.com
基于RNA-Seq技术的蜡蚧轮枝菌转录组分析
张 某, 高 熹*, 郝凯强, 谭 清, 庞仁乙, 兰明先, 李召波, 夏 涛, 鲁武锋, 李建一, 吴国星*
云南农业大学植物保护学院,云南 昆明 650201
【目的】蜡蚧轮枝菌是一种防治植物病原物和害虫的生防菌,弄清其致病的分子基础具有重要的意义。【方法】利用Illumina HiSeq技术测序蜡蚧轮枝菌的cDNA文库,并进行RNA-Seq序列拼接和功能注释。【结果】共获得1634670条高品质reads,碱基GC含量48.50%。14856条Unigene经组装拼接后,在Nr、Swiss-Prot、COG和KEGG数据库中分别比对得到1467、9379、6518和4188条Unigenes。采用GO分类工具将21403条Unigenes 分成24个功能组,主要包含在细胞组成(1369)、分子功能(1679)和生物学过程(1928)3个大类里,在COG数据库注释的基础上,把6518条Unigenes分成38个功能组,通过GO和COG 丰度识别分类,聚类的主要是一般功能预测、次生代谢产物的合成、运输和分解代谢以及信号转导机制起作用的独立基因。能与KEGG路径匹配的108条Unigenes中大部分和新陈代谢途径及次生代谢产物的合成有关。【结论】这些结果将有助于找寻蜡蚧轮枝菌的致病因子,从而丰富其致病机理。
蜡蚧轮枝菌; RNA-Seq; 转录组
bor L. LÖVEI, 郭莹)
*通信作者(Author for correspondence), 高熹, E-mail: chonchon@163.com; 吴国星, E-mail: wugx1@163.com
10. 3969/j.issn.2095-1787.2017.03.010
猜你喜欢
农业科技与信息(2022年13期)2022-11-22
国际放射医学核医学杂志(2021年6期)2021-11-30
国际放射医学核医学杂志(2021年1期)2021-11-30
山东理工大学学报(社会科学版)(2021年5期)2021-09-23
书香两岸(2020年3期)2020-06-29
中国蔬菜(2019年12期)2019-01-22
西安航空学院学报(2018年5期)2018-10-15
生物技术通报(2018年4期)2018-03-31
