
手性辅基诱导的含三氟甲砜基胺的不对称合成
2017-08-29 23:50方博杰孟卫东黄焰根东华大学化学化工与生物工程学院上海201620
合成化学 2017年8期
方博杰, 孟卫东, 黄焰根(东华大学 化学化工与生物工程学院, 上海 201620)
·快递论文·
手性辅基诱导的含三氟甲砜基胺的不对称合成
方博杰, 孟卫东, 黄焰根*
(东华大学 化学化工与生物工程学院, 上海 201620)
以(S)-叔丁基亚磺酰亚胺和苄基三氟甲基砜类化合物为原料,正丁基锂(n-BuLi)为碱,THF为溶剂, Ti(OiPr)4为路易斯酸,经不对称加成反应合成了一系列含三氟甲砜基的胺类化合物(3a~3j),产率45%~78%,d/r50 ∶26 ∶24 ∶0~85 ∶7 ∶6 ∶2,其结构经1H NMR,13C NMR,19F NMR, FT-IR和HR-MS(ESI)确证。采用X-ray单晶衍射研究了3i的构型。结果表明:3i的C1为R构型,C2为S构型。
三氟甲砜基;α-磺酰基碳负离子; 手性辅基; 不对称加成; 合成
与磺酰基相连的碳上的氢酸性较强,容易形成碳负离子。研究人员对采用α-磺酰基碳负离子构建C—C键的方法已进行了广泛的研究。α-磺酰基碳负离子很容易与各种亲电试剂反应[1]。朱仕正等[2]于1998年报道了以苄基三氟甲砜和卤代烃为原料,DMF为溶剂,K2CO3为碱,经环化反应制得三元环化合物的方法(Scheme 1)。该反应条件温和,产率较高(72%)。α-磺酰基碳负离子还常被用于合成各种天然产物[3]。

Scheme 1
部分手性胺类化合物具有良好的生物活性。如多佐胺具有抗青光眼的作用,手性胺型γ-分泌酶抑制剂对阿尔茨海默病有较好的疗效等[4]。

Scheme 2

Scheme 3
含手性磺酰基的化合物因具有类似羰基的结构和电学特性,已在药物化学领域得到了重视[5]。
探究合成光学纯的磺酰基衍生物的方法具有重要的实际意义。然而关于α-磺酰基碳负离子的不对称反应报道却较少[6]。2007年,Nakamura等[7]报道了苄基三氟甲基砜与苯甲醛在手性配体参与下合成具有良好立体选择性的加成产物(Scheme 2)。
除了利用手性配体构建手性中心外,还可以通过手性辅基诱导的方法构筑。叔丁基亚磺酰基[8]是一种常用的手性辅基,不仅具有良好的手性诱导作用,还易于脱去。
基于此,本文以苄基三氟甲基砜类化合物(1a~1j)和(S)-叔丁基亚磺酰亚胺(2)为原料,正丁基锂(n-BuLi)为碱,THF为溶剂, Ti(OiPr)4为路易斯酸,经不对称加成反应合成了一系列含三氟甲砜基的胺类化合物(3a~3j, Scheme 3),产率45%~78%,d/r50 ∶26 ∶24 ∶0~85 ∶7 ∶6 ∶2,其结构经1H NMR,13C NMR,19F NMR, FT-IR和HR-MS(ESI)确证。采用X-ray单晶衍射研究了3i的构型。结果表明:3i的C1为R构型,C2为S构型。
1 实验部分
1.1 仪器与试剂
WZZ-2S型自动旋光仪;Bruker AM-400 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Nicolet 308 FT-IR型傅里叶变换红外光谱仪(KBr压片);Thermo Fisher Scientific LTQ FT Ultra型质谱仪;Rigaku FCR Diffractimer型X-射线衍射仪。
1a~1j[2]和2[9]按文献方法合成;其余所用试剂均为分析纯。
1.2 合成
(1) 3a~3j的合成(以3a为例)
N2保护下,向干燥的反应管中加入三氟甲基苄基砜(1a)0.179 4 g(0.8 mmol)和THF 6 mL,于-78 ℃搅拌20 min;缓慢滴加n-BuLi 0.6 mL,滴毕,反应液由无色变为淡黄色,搅拌2 h;滴加2 0.117 8 g(0.8 mmol)和Ti(OiPr)40.227 4 g(0.8 mmol)的THF(1 mL)溶液,滴毕,反应12 h。加入饱和氯化铵溶液(6 mL)淬灭反应,于室温分液,水相用Et2O(2×10 mL)萃取,合并有机相,用无水硫酸钠干燥,经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=10/1,V/V)纯化得3a。
用类似的方法合成3b~3j。
(2) 4i的合成

2 结果与讨论
2.1 合成
(1) 3a的合成反应条件优化
以1a与2为模板底物,优化了反应条件,结果见表1。由表1可见,Entry 1~7为碱对产率的影响,K2CO3, KOH, DBU和LiHMDS为碱,反应无法进行,n-BuLi,t-BuLi和LDA为碱,产率较高,结合反应安全性考虑,最终选择n-BuLi为碱。Entry 6和Entry 8~12为路易斯酸对产率的影响,Ti(OiPr)4为路易斯酸,产率较高,HMPA, MgBr2, ZnBr2和(n-BuO)4Ti则对反应没有明显的促进作用。Entry 12~14为Ti(OiPr)4用量对产率的影响,当用量为1.0 eq.时,产率最高(Entry 12, 71%)。 Entry 12, Entry 15和Entry 16为底物投料比对产率的影响,当n(1a) ∶n(2)=1 ∶1时,产率最高。Entry 17~19为反应溶剂对产率的影响,以Et2O, DCM和toluene为溶剂,反应均不能进行,以THF为溶剂,产率较高。此外,我们还考查了反应温度对产率的影响(Entry 12和Entry 20~21),反应温度为-42 ℃和0 ℃时,TLC监控发现大量副产物。因此,-78 ℃为最佳反应温度。
综上,最佳反应条件为:n(1a) ∶n(2)=1 ∶1,n-BuLi(1.2 eq)为碱,Ti(OiPr)4(1.0 eq.)为路易斯酸,THF为反应溶剂,于-78 ℃反应12 h, 3a产率71%。

Scheme 4

Entry碱路易斯酸1a∶2i溶剂产率/%b1K2CO3-1∶1THF02KOH-1∶1THF03DBU-1∶1THF04LiHMDS-1∶1THF05LDA-1∶1THFtrace6n⁃BuLi-1∶1THF537t⁃BuLi-1∶1THFtrace8n⁃BuLiHMPA1∶1THFtrace9n⁃BuLiMgBr21∶1THF4810n⁃BuLiZnBr21∶1THF1511n⁃BuLi(n⁃BuO)4Ti1∶1THF1612n⁃BuLiTi(OiPr)41∶1THF71(65)c13n⁃BuLiTi(OiPr)4g1∶1THF1814n⁃BuLiTi(OiPr)4h1∶1THF2115n⁃BuLiTi(OiPr)41∶2THF6116n⁃BuLiTi(OiPr)42∶1THF5917n⁃BuLiTi(OiPr)41∶1Et2O018n⁃BuLiTi(OiPr)41∶1DCM019n⁃BuLiTi(OiPr)41∶1toluene020en⁃BuLiTi(OiPr)41∶1THFNDd21fn⁃BuLiTi(OiPr)41∶1THFNDd
a碱1.2 eq., 路易斯酸1.0 eq., 于-78 ℃反应过夜;b由19F NMR分析得到;c分离收率;d未检测出;e反应温度为-42 ℃;f反应温度为0 ℃;g0.5 eq.;h1.5 eq.;i摩尔比。
(2) 反应普适性
在最优化反应条件下,我们对反应底物的普适性进行了研究(Scheme 3)。结果表明,底物1中苯环上的取代基对产率有较大影响。当苯环上有苯基取代或底物为萘环取代时,产率中等(3b和3c),分别为62%和45%。当苯环上对位或间位有给电子基时,产率提高(3d, 3e和3f),最高产率为78%。苯环对位上为卤素取代时,产率下降(3h)。苯环上为3,5-二取代的给电子基时,产率也较高(3i, 61%和3j, 59%)。苯环上的取代基对选择性也有一定影响,当苯环上有位阻较大的基团或含有卤素时,选择性下降(3b, 3c和3h)。含有3,5-二取代给电子基团时,选择性也较低(3j)。
(3) 脱保护
3i可以在酸性条件下脱去叔丁基亚磺酰基,并以92%的产率合成铵盐产物4i(Scheme 4)。需要注意的是,3i脱除保护基后,若用碱中和反应液,所得产物为氨基消除的烯烃化合物5。这可能是由于三氟甲磺酰基的强吸电子性使得苄位的氢很容易被消除。
2.2 表征
图1为3i的单晶衍射结构。由图1可见,C2为S构型,C1为R构型,综合19F NMR结果,我们推测其它产物的绝对构型与3i一致。

图1 3i的单晶衍射结构
2.3 反应机理
根据产物的绝对构型,对反应机理进行了推测(Scheme 5)。 1a在n-BuLi作用下脱除苄基三氟甲基砜苄位的氢,形成离子A。2与Lewis酸Ti(OiPr)4络合形成活化中间体B。A从位阻较小的一面进攻B得到加成产物。
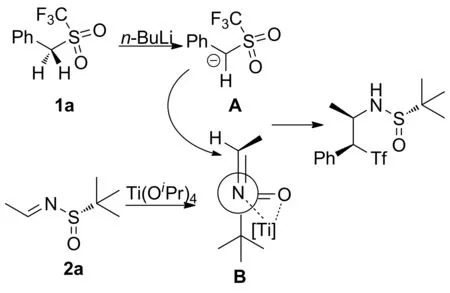
Scheme 5
采用苄基三氟甲砜与(S)-叔丁基亚磺酰亚胺的不对称加成反应合成了含三氟甲砜基胺类化合物,产率较高,选择性中等。该反应体系原料易得,条件温和,操作简单,为含三氟甲砜基的胺类化合物的合成提供了借鉴。
[1] Blackmore P R. The modified Julia olefination:Alkene synthesisviathe condensation of metallated heteroarylalkylsulfones with carbonyl compounds[J].J Chem Soc Perkin Trans,2002,1:2563-2585.
[2] Zhu S Z, Chu Q, Xu G L,etal. A convenient one-pot synthesis of per-(or poly-) fluoroalkanesulfonyl substituted cyclopropanes[J].J Fluorine Chem,1998,91:195-198.
[3] Evans D A, Dow R L, Shih T L,etal. Total synthesis of the polyether antibiotic ionomycin[J].J Am Chem Soc,1990,112:5290-5313.
[4] Nakamura S, Hirata N, Yamada R,etal. Catalytic and highly enantioselective reactions ofα-sulfonyl carbanions with chiral bis(oxazoline)s[J].Chem Eur J,2008,14:5519-5527.
[5] Tsang W Y, Ahmed N, Harding L P,etal. Acylation versus sulfonylation in the inhibition of elastase by 3-oxo-β-sultams[J].J Am Chem Soc,2005,127:8946-8947.
[6] Arai S, Ishida T, Shioiri T. Asymmetric synthesis ofα,β-epoxysulfones under phase-transfer catalyzed Darzens reaction[J].Tetrahedron Lett,1998,39:8299-8302.
[7] Nakamura S, Hirata N, Kita T,etal. Highly enantioselective reactions ofα-sulfonyl carbanions of trifluoromethyl sulfones[J].Angew Chem Int Ed,2007,46:7648-7650.
[8] Robak M T, Herbage M A, Ellman J A. Synthesis and applications oftert-butanesulfinamide[J].Chem Rev,2010,110:3600-3740.
[9] Collados J F, Toledano E, Guijarro D,etal. Microwave-assisted solvent-free synthesis of enantiomerically pureN-(tert-butylsulfinyl)imines[J].J Org Chem,2012,77:5744-5750.
Chiral Auxiliary Induced Asymmetric Synthesis ofAmines Containing Triflyl Group
FANG Bo-jie, MENG Wei-dong, HUANG Yan-gen*
(College of Chemistry, Chemical Engineering and Biotechnology, Donghua University, Shanghai 201620, China)
A series of amines(3a~3j) containing triflyl group were synthesized by asymmetric addition reaction, using (S)-tert-butylsulfoximine and aryl trifluoromethyl sulfone as starting materials,n-BuLi as base, THF as solvent and Ti(OiPr)4as Lewis acid. The yields andd/rwere 45%~78% and 50 ∶26 ∶24 ∶0~85 ∶7 ∶6 ∶2, respectively. The structures were confirmed by1H NMR,13C NMR,19F NMR, FT-IR and HR-MS(ESI). The configuration of 3i was investigated by X-ray single crystal diffraction. The results indicated that the configuration of C1 and C2 wereRandS, respectively.
triflyl;α-sulfonyl carbanion; chiral auxiliary; asymmetric addition; synthesis
2017-01-23;
2017-06-26
上海市教委科研创新项目(13ZZ047)
方博杰(1990-),男,汉族,湖北宜昌人,硕士研究生,主要从事含氟胺类化合物的不对称合成的研究。 E-mail: fangbojie@yeah.net
黄焰根,副教授, E-mail: hyg@dhu.edu.cn
O623.7; O622.6
A
10.15952/j.cnki.cjsc.1005-1511.2017.08.17016
猜你喜欢
中学化学(2022年5期)2022-06-17
华南师范大学学报(自然科学版)(2021年4期)2021-08-30
食品安全导刊(2021年21期)2021-08-30
理科考试研究·高中(2019年8期)2019-09-19
中成药(2018年7期)2018-08-04
中国学术期刊文摘(2016年8期)2016-02-13
化学教学(2015年11期)2015-12-19
中国洗涤用品工业(2015年9期)2015-02-28
中国洗涤用品工业(2015年2期)2015-02-28
中国塑料(2014年10期)2014-10-17
