
磁性石墨烯固相萃取—高分辨质谱法快速测定动物源性食品中磺胺类药物残留
2017-07-15 02:55崔晓娜葛爱民李汝春董彦莉
江苏农业科学 2017年9期
关键词:磺胺
崔晓娜++葛爱民++李汝春++董彦莉

摘要:采用高效液相色谱-静电场轨道肼高分辨质谱技术,建立了动物源性食品中13种磺胺类药物残留的磁性石墨烯固相萃取-高分辨质谱分析方法。样品采用0.1%的甲酸/乙腈提取,目标物通过磁性石墨烯粉吸附,除去杂质,然后再解析目标物的方法获得干净的样品基质。经Waters UPLC BEH C18(2.1 mm×100 mm,1.7 μm)色谱柱分离,液相色谱-高分辨质谱仪进行检测。结果表明,13种目标物在2.0~50.0 μg/L范围内线性关系良好(r2>0.99)。在动物源性食品基质中,目标化合物在2、5、10 μg/kg 3个加标水平的平均回收率在77.8%~98.4%,相对标准偏差(RSD)为4.1%~10.1%。该方法净化速度快、效率高,可用于动物源性食品中磺胺类药物残留筛查检测。
关键词:磁性石墨烯固相萃取(MSPE);高分辨质谱法;动物源性食品;磺胺
中图分类号: TS207文献标志码: A
文章编号:1002-1302(2017)09-0151-04
磺胺类药物为合成的广谱抗菌剂,对多数革兰氏阳性菌和阴性菌有效。由于磺胺类药物在体内作用时间和代谢时间较长,通过任何途径摄入的磺胺都有可能在人体中蓄积,蓄积到一定值的药物对人体机能是有害的[1]。目前,我国对磺胺类及其增效剂的使用有比较明确的规定。我国农业部第235号公告中规定,禽肉类产品中磺胺类总量不得超过 0.1 mg/kg;日本肯定列表中将动物源性食品的最低限量定为 0.02 mg/kg;欧盟2377/90/EEC中明确规定,在所有动物源性食品中磺胺类残留总量不能超过0.1 mg/kg[2-3]。
磁性石墨烯是由石墨烯化学制备而成,磁性石墨烯纳米粒子因具有很多特殊的磁性质,比如超顺磁性、高矫顽力、低居里温度、高磁化率等,并且在磁流体、数据存储、催化、污染物处理等很多方面都已有广泛的研究和实际应用。所以石墨烯与磁性纳米粒子复合成的磁性石墨烯受到了极大的关注,并为石墨烯的应用研究发展出了新的方向[4-6]。
目前,磺胺类及其增效剂残留的测定方法有高效液相色谱法配合紫外检测器、荧光检测器和质谱检测器[7]。液相色谱-三重四极杆串联质谱(LC/MS/MS)可以在一次分析中定量和鉴定数百种目标化合物,具有很高的选择性和灵敏度。但是,这种方法需要采用多种已知化合物优化仪器参数,因此不能用于筛查未知化合物。基于Orbitrap技术,Exactive 具有极高的质量分辨率,从而保证了分析结果高质量准确度,并可以準确地区分复杂样品中同时洗脱出的具有相同质量数的化合物,高达3个数量级的动态线性范围,可轻松分析受复杂基体干扰的痕量化合物[8-10]。
本研究建立了动物源性食品中13种具有代表性的磺胺类药物增效剂的快速前处理和高分辨质谱法联用的测定方法,该方法快速、准确,为监控动物源性食品中磺胺类药物增效剂残留提供了技术支持。
1材料与方法
1.1试剂与材料
1.1.1标准物质磺胺嘧啶、磺胺噻唑、磺胺吡啶、磺胺甲基嘧啶、磺胺二甲基嘧啶、磺胺-5-(对)甲氧嘧啶、磺胺甲噻二唑、磺胺甲氧哒嗪、磺胺氯哒嗪、磺胺-6-(间)甲氧嘧啶、磺胺邻二甲氧嘧啶、磺胺甲基异[XCZ1.tif]唑、磺胺喹[XCZ1.tif]啉,均购自DR.公司,纯度均大于99%。
1.1.2试剂与耗材乙腈,甲醇均为色谱纯(Fisher);无水硫酸钠,分析纯(烟台市双双化工);甲酸,色谱纯(TEDIA);水,GB/T 6682规定的一级水;磁性石墨烯,实验室制备;硫酸铜,优级纯(天津市北联精细化学品开发有限公司);1-(2-吡咯偶氮)-2-萘酚(PAN),分析纯(北京化学试剂有限公司);C18粉(天津艾杰尔)。
1.1.3仪器液相色谱仪,Thermo UltiMate3000,静电场轨道肼质谱仪(Thermo EXACTIVE);分析天平,感量0.1 mg和0.01 g各1台;离心机,5810型(Eppendorf公司);均质器(T25型,IKA公司);振荡器(MMV-1000W,ETELA公司);氮吹仪(N-EVAP);涡流混匀器(IKA MS basic)。
1.1.4标准溶液配制
磺胺嘧啶、磺胺噻唑、磺胺吡啶、磺胺甲基嘧啶、磺胺二甲基嘧啶、磺胺-5-(对)甲氧嘧啶、磺胺甲噻二唑、磺胺甲氧哒嗪、磺胺氯哒嗪、磺胺-6-(间)甲氧嘧啶、磺胺邻二甲氧嘧啶、磺胺甲基异[XCZ1.tif]唑、磺胺喹[XCZ1.tif]啉:用乙腈溶解,按纯度标示折算配制成100 μg/mL的标准储备液。
1.2试验方法
1.2.1样品提取
准确称取2.00(±0.02)g待测样品置于100 mL离心管中,加入5 g无水硫酸钠,2 g氯化钠,10 mL 0.1% 甲酸/乙腈,均质2 min,10 000 r/min离心10 min,将上清液转入另一干净离心试管中,重复提取残渣1次,取上清液合并,加入10 mL正己烷,振荡5 min后静置,去掉上层正己烷层。将提取液旋转蒸发至干,加入3 mL乙腈/水溶解,待净化。
1.2.2样品净化
准确称取20 mg磁性石墨烯于10 mL离心管中,分别加入2 mL丙酮、甲醇和去离子水,依次清洗磁性石墨烯。取5 mL上清液入离心管中,涡混10 min,静止萃取5 min。用磁铁将磁性石墨烯聚集到离心管底部,弃去上清液,将磁性石墨烯转移至另1个干净的离心试管中,加入 1 mL 乙腈-氨水(体积比为95 ∶5),涡旋2 min洗脱,重复洗脱4次,合并洗脱液,氮气吹干,流动相定容至1 mL,过 0.22 μm 滤膜,进行质谱测定分析。
1.2.3液相色谱条件
色谱柱:Waters UPLC BEH C18(2.1×100 mm,1.7 μm);柱温:40 ℃,流速:0.3 mL/min,进样量:20 μL;流动相:A:0.1%甲酸水,B:甲醇,梯度洗脱程序:0→2.0 min,由5% B→20% B;2.0→6 min,由20% B→80% B;6.2 min,5% B,平衡3 min。
1.2.4质谱条件
全扫描正离子模式(质量范围100~1 500);分辨率:50 000;自动增益控制(AGC)目标值:10e6;加热点喷雾离子源条件:喷雾电压:2 200 V,离子传输管温度:280 ℃,鞘气气压:32 au,气化室温度:200 ℃。13种化合物的质谱参数详见表1。
2结果与分析
2.1质谱条件的优化
2.1.1色谱柱的选择
本研究检测的13种磺胺类药物中磺胺-5-(对)甲氧嘧啶、磺胺甲氧哒嗪、磺胺-6-(间)甲氧嘧啶分子量相同,因此,不同目标物的分离要考虑化合物性质,在提高分离速度和灵敏度的同时,达到最佳的目标物分离效果。通过比较Waters UPLC BEH C18(粒径1.7 μm)和Agilent ZORBAX Eclipse Plus C18(粒径1.8 μm),以及柱长分别为2.1×100 mm、3.0×100 mm的2种不同规格的色谱柱,在分离效率和目标物响应值方面,柱长为2.1×100 mm Waters UPLC BEH C18更好一些。
2.1.2流动相的选择
磺胺类药物在正离子模式下响应值较高,通过在水溶液中加入酸性溶液的方式测试其响应值。分别比较了甲醇/0.1%甲酸水溶液、乙腈/0.1%甲酸水、乙腈/10 mmol/L乙酸铵、甲醇/10 mmol/L乙酸铵溶液作为流动相,在电喷雾源ESI+方式下,在甲醇/0.1%甲酸水溶液组成的流动相中,质谱的离子化效率较其他流动相条件响应值高,峰形好,因此本研究选择甲醇/0.1%甲酸水作为流动相。
通过改变甲醇/0.1%甲酸水不同的起始比例,经多次进样分离,结果表明,在采用梯度洗脱的条件下,起始流动相甲醇比例在5%时,磺胺-5-(对)甲氧嘧啶、磺胺甲氧哒嗪、磺胺-6-(间)甲氧嘧啶能够得到较好地分离。在本研究的色谱条件下,13种化合物的出峰情况见图1。
2.2前处理条件的优化与比较
2.2.1提取试剂的选择
磺胺类药物其结构中均含有多个胺基团,在酸性条件下容易形成正离子,因此,在提取试剂中加入酸性溶液,可以提高提取效率和离子化效果[11]。在提取试剂选择方面,分别比较了0.1%的甲酸乙腈、0.1%的甲酸甲醇、0.1%的甲酸乙酸乙酯的提取效率,结果表明,0.1%的甲酸乙腈提取效率较高,同时加入2 g氯化钠能够使乙腈层和水层较好地分离,避免乳化现象出现。
2.2.2净化吸附剂的优化
QuEChERS(分散固相萃取)方法的原理是通过吸附粉末的吸附作用而达到净化的效果,此种净化方法主要去除的是极性中小分子杂质,而液质联用残留分析中主要用到的电喷雾电离源(ESI源)分析的就是极性化合物,因此二者相互结合,可以发挥最大的检测效率[12-13]。利用质谱在全扫描方式下对ODSC18和MSPE净化效果进行考察,从图2可以看出,MSPE方法净化完后一些中极性和分子量较大的杂质被除去,综合比较,在杂质净化方面MSPE要优于C18。
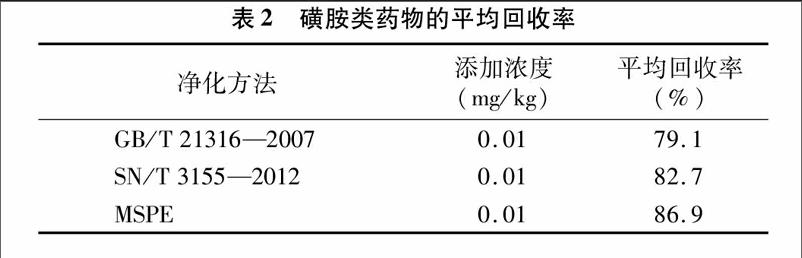
2.3磁性石墨烯净化方法与国标、行标的比较
在GB/T 21316—2007中前处理净化采用的是液液萃取方法,SN/T 3155—2012中肉及肉制品的净化采用的是过C18固相萃取柱的方法[14-15],根据3种前处理方法,分别添加质量浓度为0.01 mg/kg的标准物质在空白样品当中,磺胺类药物的平均回收率见表2。
2.4方法的线性、精密度、检测限和定量限
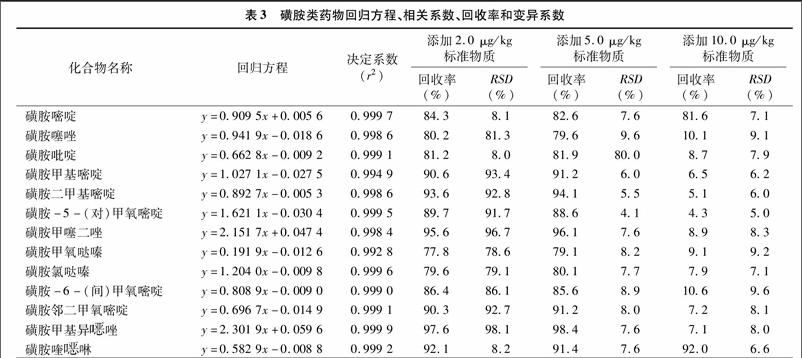
考察了试验方法及仪器條件的线性及精密度,选取不同的[CM(25]动物源性食品,分别添加质量浓度为[KG*5]2.0、5.0、10.0 μg/kg 的标准物质,线性回归方程、相关系数、回收率、RSD值见表3。
3结论
本研究采用高效液相色谱-静电场轨道肼高分辨质谱,磁性石墨烯净化技术,建立了动物源性食品中磺胺类药物残留的液相色谱-高分辨质谱分析检测方法。采用磁性石墨烯固相萃取作为净化材料,提高了样品前处理效率,降低了环境污染,为监控动物源性食品中磺胺类药物增效剂残留提供了有力的技术支持。
参考文献:
[1]沈建忠,谢联金. 兽医药理学[M]. 北京:中国农业大学出版社,2000.
[2]农业部. 动物性食品中兽药最高残留限量(农业部2002年235号公告)[J]. 中国猪业,2010(8):10-12.
[3]蒋原,沈崇钰,姚义刚,等. 动物源性食品中磺胺类药物增效剂残留的高效液相色谱-串联质谱法测定[J]. 分析测试学报,2009,28(7):834-837.
[4]Liu Q,Shi J B,Zeng L X,et al. Evaluation of graphene as an advantageous adsorbent for solid-phase extraction with chlorophenols as model analytes[J]. Journal of Chromatography A,2011,1218(2):197-204.
[5]Hummers W,Offeman R. Preparation of graphite oxide[J]. Journal of American Chemical Society,1958,80(6):1339.
[6]Luo Y B,Shi Z G,Gao Q,et al. Magnetic retrieval of graphene:extraction of sulfonamide antibiotics from environmental water samples[J]. Journal of Chromatography A,2011,1218(10):1353-1358.
[7]庞国芳. 农药兽药残留现代分析技术[M].北京:科学出版社,2007:366-367.[HJ1.8mm]
[8]Jia Q N,Zhao G C. Preparation of graphene-based solid phase microextraction fiber and its determination of polychlorinated biphenyls[J]. Journal of Instrumental Analysis,2015,32(5):541-546.
[9]李佐卿,倪梅林,俞雪钧,等. 液相色谱-串联质谱法检测水产品中磺胺类和喹诺酮类药物残留[J]. 分析测试学报,2007,26(4):508-510,514.
[10]杨艳伟,朱英. HPLC法测定化妆品中11种磺胺类化合物[J]. 中国卫生检验杂志,2006,16(11):1343-1344.
[11]宁啸骏,王丁林,虞成华,等. UPLC-MS/MS同位素内标法测定食品中对位红、苏丹红Ⅰ~Ⅳ的研究[J]. 质谱学报,2009,30(1):41-46.
[12]Fischer J,Kelly M T,Smyth M R,et al. Determination of ivermectin in bovine plasma by column-switching LC using on-line solid-phase extraction and trace enrichment [J]. Journal of Pharmaceutical & Biomedical Analysis,1993,11(3):217-223.
[13] Mckellar Q A,Benchaoui H A. Avermectins and milbemycins[J]. Journal of Veterinary Pharmacology & Therapeutics,1996,19(5):331-351.
[14]国家质量监督检验检疫总局. 动物源性食品中磺胺类药物残留量的测定液相色谱-质谱/质谱法:GB/T 21316—2007[S]. 北京:中国标准出版社,2007.
[15]国家质量监督检验检疫总局. 出口猪肉、虾、蜂蜜中多种药物残留量的测定:SN/T 3155—2012[S]. 北京:中國标准出版社,2012.
猜你喜欢
中国饲料(2022年5期)2022-04-26
中兽医学杂志(2021年1期)2021-03-27
武警医学(2018年10期)2018-11-06
环境科技(2016年4期)2016-11-08
山东畜牧兽医(2016年2期)2016-04-05
湖南农业(2016年12期)2016-03-10
现代农业(2016年4期)2016-02-28
应用化工(2015年1期)2015-12-24
环境科技(2015年6期)2015-11-08
无机化学学报(2014年3期)2014-02-28
