
共聚酰胺6/66的合成与表征
2017-06-27 08:10:01罗玉航易春旺
合成纤维工业 2017年3期
罗玉航,彭 露,姜 锋,张 维,易春旺,3*
(1.湖南师范大学 资源精细化与先进材料湖南省高校重点实验室,湖南 长沙 410081;2.中国纺织科学研究院生物源纤维制造技术国家重点实验室,北京 100025; 3.湖南师范大学 石化新材料与资源精细利用国家地方联合工程实验室,湖南 长沙 410081)
共聚酰胺6/66的合成与表征
罗玉航1,2,彭 露1,姜 锋2,张 维1,易春旺1,3*
(1.湖南师范大学 资源精细化与先进材料湖南省高校重点实验室,湖南 长沙 410081;2.中国纺织科学研究院生物源纤维制造技术国家重点实验室,北京 100025; 3.湖南师范大学 石化新材料与资源精细利用国家地方联合工程实验室,湖南 长沙 410081)
采用己内酰胺和己二酸己二胺盐等为原料合成系列共聚酰胺6/66,研究了共聚单体配比、开环剂及相对分子质量调节剂对共聚物性能的影响。通过傅里叶变换红外光谱、核磁共振氢谱、X射线衍射和差示扫描量热仪分别对共聚物的结构与热性能进行了测试。结果表明:己二酸己二胺盐和ε-氨基己酸对共聚反应不仅具有一定的促进作用,还降低了共聚物的可萃取物含量;己二酸己二胺盐的加入并未改变主体聚酰胺6的晶型结构,均为稳定的α晶型;共聚酰胺6/66的熔融双峰是由于晶体结构的重组导致的。
聚己内酰胺 聚己二酸己二胺 共聚酰胺 合成 晶体结构 结构表征
共聚酰胺是由两种或两种以上的聚酰胺单体或缩聚物进行聚合得到的聚酰胺产品[1]。通过改变单体配方和配比可以制备出性能各异的共聚酰胺,从而实现聚酰胺的高性能化和功能化,并进一步扩大其应用领域。
共聚酰胺6/66具有低熔点、低结晶度等特点,且阻隔性能、耐磨性、耐溶剂性等优异,备受研究者的关注[2-4]。K.Suehir等[5]着重研究了共聚酰胺6/66不规则化学结构对其熔点的影响,结果表明共聚酰胺6/66的熔点-组成曲线呈V形。E.D.Harvey[4]等采用偏振光方法,研究了无规共聚物共聚酰胺66/6的结晶速率问题。
目前,国内外对共聚酰胺6/66的研究主要集中在其熔点、结晶及应用等方面,而针对配方及共聚工艺的研究相对较少。共聚酰胺6/66的制备在国外已经取得突破性进展,并已经成功商业化,但国内还处于摸索阶段。作者通过改变己内酰胺(CPL)和己二酸己二胺盐(AH-salt)的配比,制备了系列共聚酰胺6/66,研究了CPL和AH-salt的配比、配方对产物性能的影响,以期为共聚酰胺6/66的研究提供一些具有价值的理论数据。
1 实验
1.1 原料及试剂
CPL:工业级,中国石化巴陵石化分公司产;AH-salt:工业级,德国BASF公司产;氘代三氟乙酸、ε-氨基己酸:分析纯,美国Aladdin公司产;精对苯二甲酸(PTA):化学纯,北京化工厂产;浓硫酸:分析纯,质量分数为96%~98%,北京化工厂产。
1.2 共聚酰胺6/66的制备
将干燥好的原料按一定配比加入到250 mL圆底四口烧瓶中,在氮气气氛下,升温到250 ℃,反应8 h出料。不同原料配比得到的产物共聚酰胺6/66试样见表1,产物经粉碎密封保存。
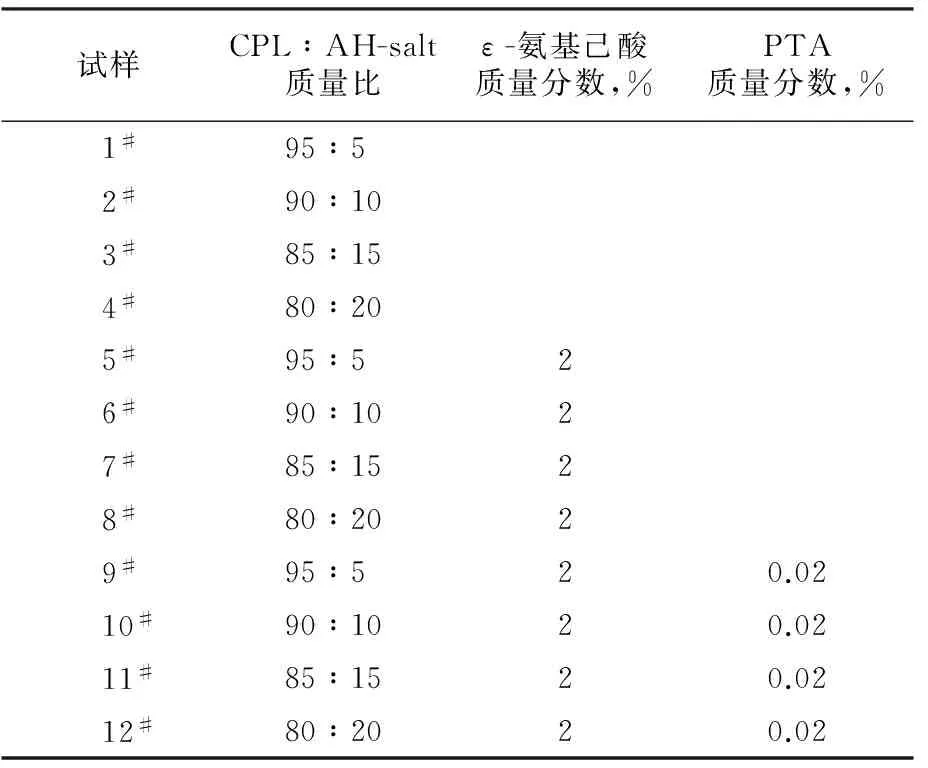
表1 共聚体系所加入的反应物的配比Tab.1 Reactants proportion of polymerization system
1.3 分析测试
可萃取物含量(W):将一定质量的试样放入真空烘箱中,在60 ℃下干燥12 h,然后称取干燥后的试样质量(m1)(精确到0.000 1 g),置于索氏提取器中,加热回流,沸水萃取12 h。萃取后的试样待冷却并自然风干其表面水后再在85 ℃下真空干燥12 h,冷却后再次称量其质量(m2),根据式(1)即可计算得到试样的W。
W=(m1-m2)/m1×100%
(1)
相对黏度(ηr):称取0.25g(精确到0.000 1g)试样,溶解在25mL质量分数为96%的浓硫酸中,配制成0.01g/mL溶液。在(25±0.02)℃恒温水浴中测试液体流经黏度计玻璃球两刻度线的时间,反复测定3次,3次测定值相差不超过0.1s,取其平均值。按式(2)计算ηr:
ηr=t2/t1
(2)
式中:t1为纯溶剂流出的时间;t2为聚合物溶液流出的时间。
核磁共振氢谱(1H-NMR)分析:采用德国Bruker公司的AvanceIII400型核磁共振仪在室温下进行测试,以氘代三氟乙酸作为溶剂,频率为400MHz。
傅里叶变换红外光谱(FTIR)分析:采用北京瑞利分析仪器公司的WQF-200型傅里叶变换红外光谱仪进行测试,扫描波数500~ 4 000cm-1。
差示扫描量热(DSC)分析:采用美国PerkinElmer公司的DSC8000型仪器进行测试。在氮气气氛下,升降温速率均为20 ℃/min。先从30 ℃升温到250 ℃,恒温5min,消除热历史;再由250℃降温到30 ℃,恒温5min;然后重新从30 ℃升温到240 ℃。
X射线衍射(XRD)分析:采用荷兰帕纳科公司的X′PertProMPDX射线仪进行表征,Cu靶,扫描速度为5(°)/min,扫描范围为5°~40°。
2 结果与讨论
2.1 原料配比对共聚酰胺6/66的W和ηr影响
从表2可知:1#~4#共聚酰胺6/66的W随着AH-salt添加量的增加而有所减少,但并不十分明显;但加入开环剂ε-氨基己酸后(5#~12#试样),共聚酰胺6/66的W随着AH-salt添加量的增加而明显降低,尤其在AH-salt的质量分数达到15%~20%后,共聚酰胺6/66的W降低非常明显,最低为5.47%(8#试样),说明共聚单体转化率较高。同时,ε-氨基己酸的加入对共聚酰胺6/66相对分子质量的增加有明显的促进作用,表现为产物的ηr提高显著,当AH-salt的质量分数达到20%时,共聚物ηr下降非常明显,这是因为在高温聚合条件下,AH-salt会发生分解,加入量越大,分解越多,且部分己二胺会挥发(表现为回流量加大,聚合温度下降),导致配比变化,反应体系中多余的己二酸起到了链终止剂的作用,从而使得共聚物ηr下降,这也是常压共聚的局限性。
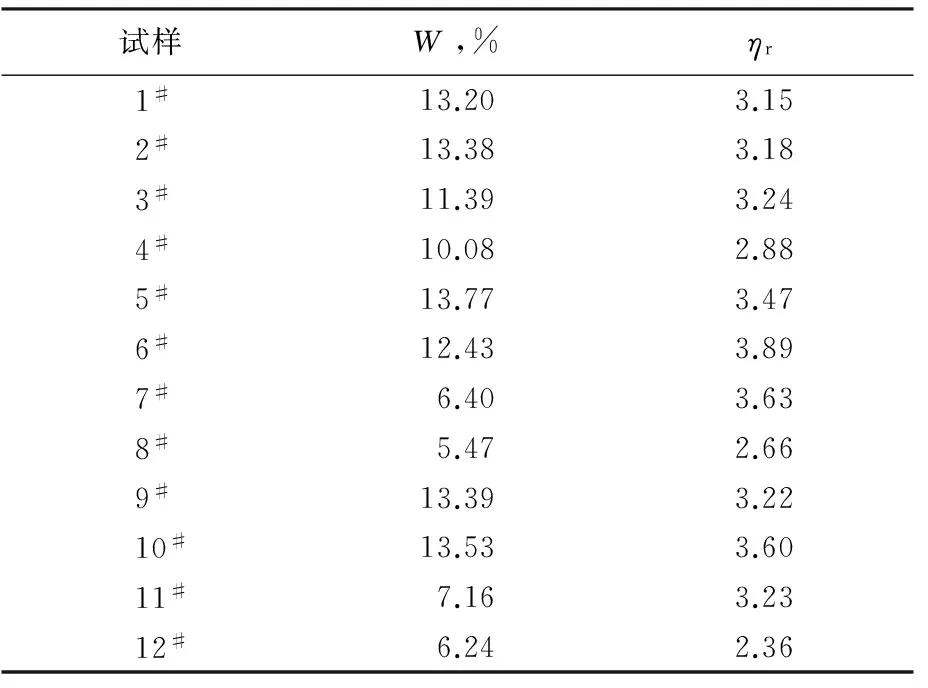
表2 共聚酰胺6/66试样的W及ηrTab.2 W and ηr of copolyamide 6/66 samples
另外,从表2还可以看出,相对分子质量调节剂PTA的加入对W的影响并不明显,但是却对相对分子质量的增长起到明显抑制作用。在CPL与AH-salt配比相同时,当加入适量的PTA后,共聚物的ηr明显下降,这说明PTA有效封闭了体系中的部分端氨基,控制了共聚酰胺6/66的相对分子质量,从而使其ηr降低。
2.2 共聚酰胺6/66的FTIR分析
由图1可知:9#~12#试样呈现出了典型的聚酰胺红外特征峰;在波数3 293 cm-1处为胺基(—NH2)的伸缩振动吸收峰;2 934 cm-1处为亚甲基(—CH2—)的不对称伸缩振动吸收峰;2 864 cm-1处为亚甲基(—CH2—)的对称伸缩振动吸收峰;1 634 cm-1处为酰胺Ⅰ的特征吸收峰;1 537 cm-1处为酰胺Ⅱ的特征吸收峰。N.Vasanthan[6]和A.Okada[7]等研究表明聚酰胺6的不同晶型在红外光谱的1 400~800 cm-1会出现相应的特征峰,其中出现在928 cm-1和1 199 cm-1两处的吸收峰归属于α晶型,因此可以采用FTIR进行鉴别,而图1试样的FTIR中均出现了上述α晶型特征吸收峰,说明共聚酰胺6/66试样中存在稳定的α晶型。
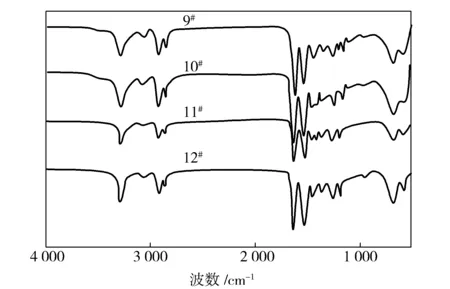
图1 共聚酰胺6/66试样的FTIRFig.1 FTIR spectra of copolyamide 6/66 samples
2.3 共聚酰胺6/66的1H-NMR分析
由图2可知,共聚物中的氢原子根据化学环境的不同,大致可以分为5类:化学位移(δ)3.651处为共聚酰胺6/66链段中胺基的α位亚甲基质子的吸收峰;δ2.828处为共聚物链段中羧基的α位亚甲基质子的吸收峰;δ1.925处是共聚物链段中胺基β位亚甲基质子的吸收峰;δ1.843处是共聚物链段中羧基β位亚甲基质子的吸收峰;δ1.604处是共聚物链段中胺基γ位亚甲基质子的吸收峰,并未出现杂质峰,各类氢原子都有归属,且各部分的积分面积比与各类氢原子的个数比相符。因此,结合共聚酰胺6/66的FTIR,说明成功合成了共聚酰胺6/66。
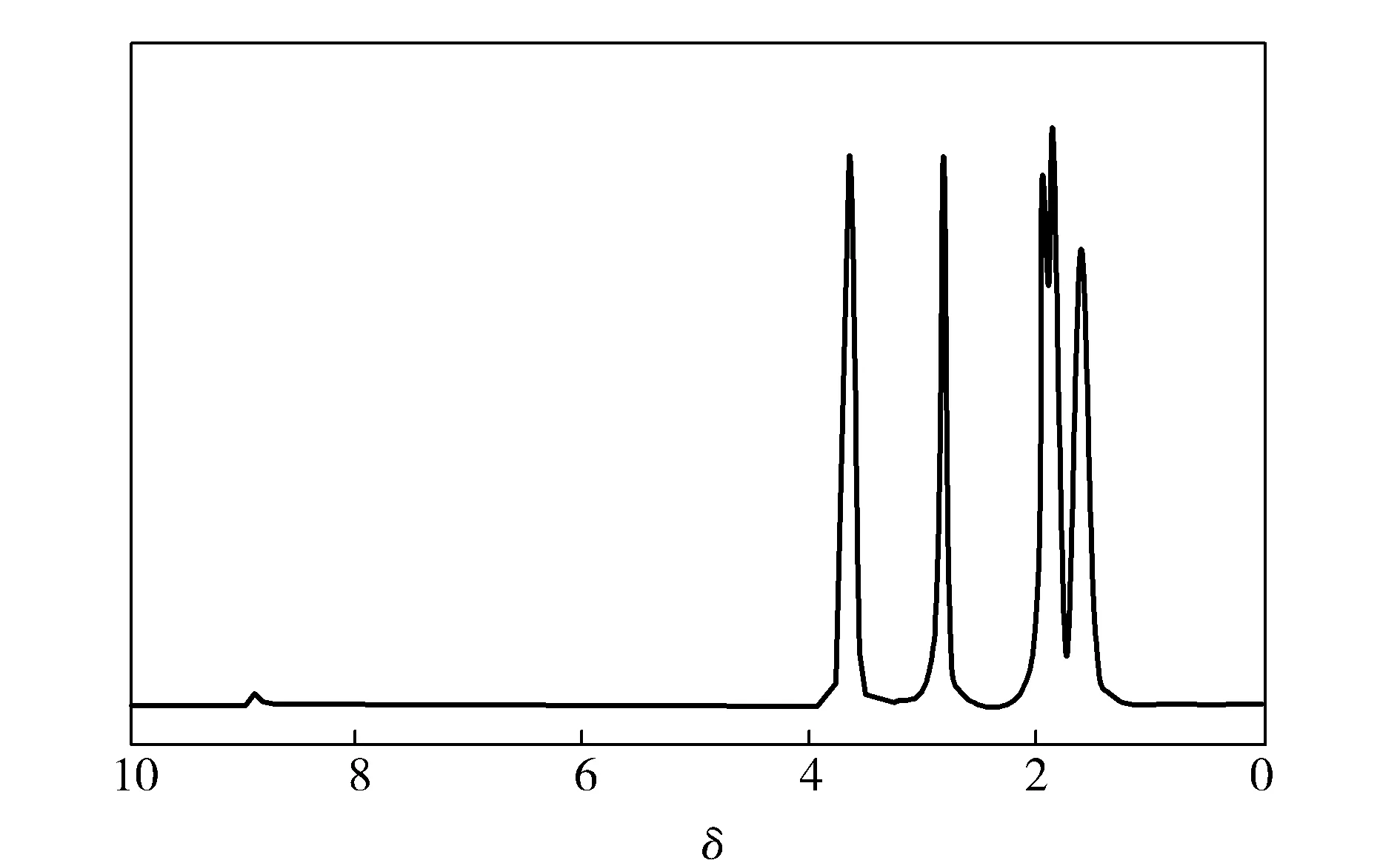
图2 11#试样的1H-NMRFig.2 1H-NMR spectra of sample 11#
2.4 共聚酰胺6/66的XRD分析
聚酰胺6存在α,β和γ3种晶型,其中β晶型为不稳定晶型,γ晶型为亚稳定晶型,α晶型的2θ约在20.2°和23°出现特征衍射峰,γ晶型的2θ约在11°和21.3°出现特征衍射峰[8]。聚酰胺66通常存在α,β两种晶型,α晶型的2θ约在20.3°和23.5°出现特征衍射峰,β晶型的2θ约在13.5°和21.3°出现特征衍射峰。由图3可知,试样在20.5°和24.4°左右呈现出特征衍射峰,说明在该聚合实验条件下所制备的共聚酰胺6/66为稳定的α晶型,这与FTIR分析结果相符。

图3 共聚酰胺6/66试样的XRD光谱Fig.3 XRD spectra of copolyamide 6/66 samples
2.5 共聚酰胺6/66的DSC分析
受熔融重结晶、不同的结晶结构、不同尺寸大小的晶体、退火产生新的二次结晶的晶体等因素影响,半结晶聚合物的DSC曲线中大多出现熔融多峰[9]。由图4可知,共聚酰胺6/66试样的升温曲线出现了熔融双峰,结合FTIR和XRD的分析结果,可以证明共聚酰胺6/66的熔融双峰不是因为存在多种晶型的影响,而是由于晶体结构的重组[10]。
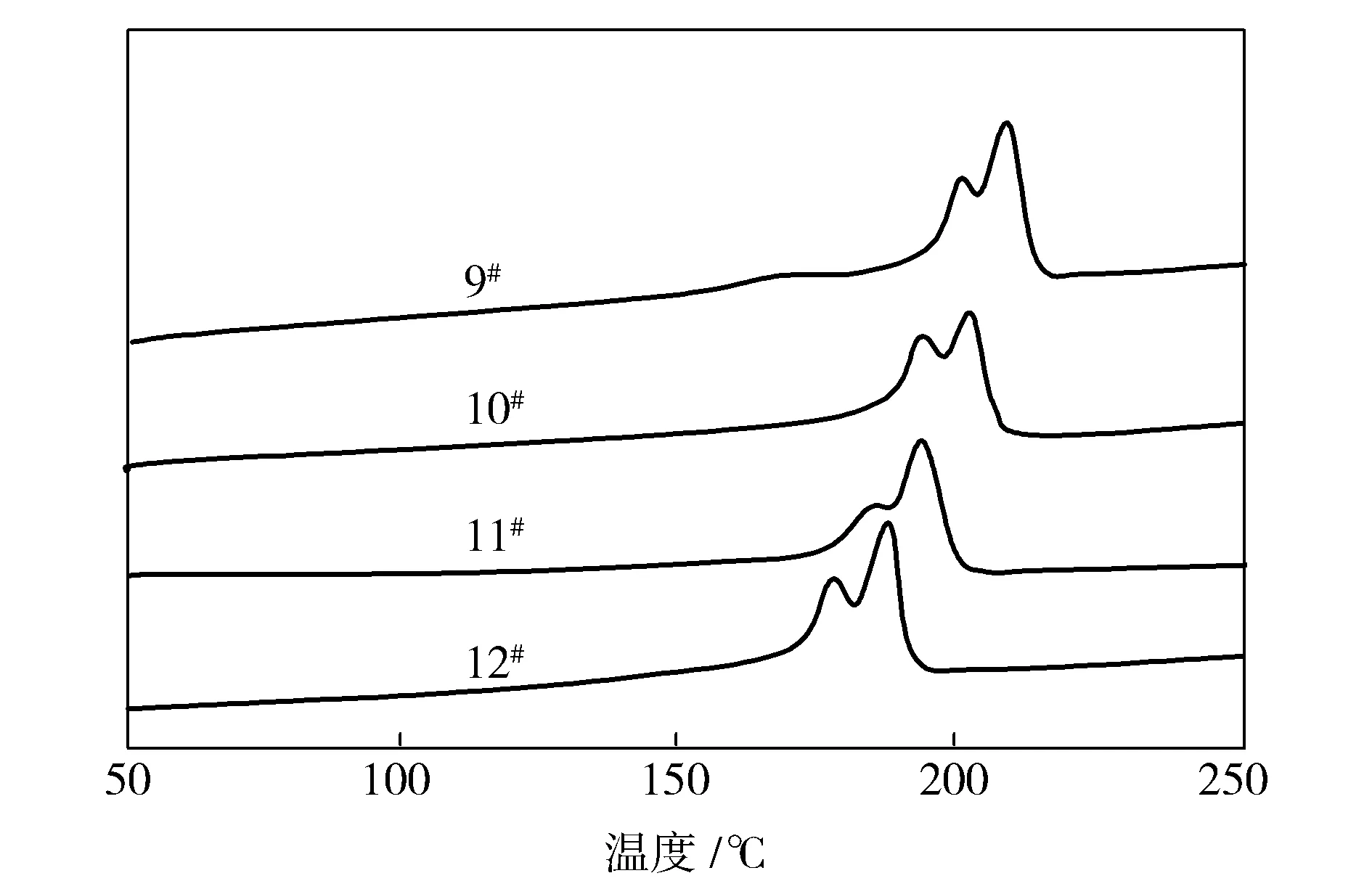
图4 共聚酰胺6/66试样的DSC曲线Fig.4 DSC curves of copolyamide 6/66 samples
由表3可知,随着AH-salt添加量的增加,共聚酰胺6/66的熔融温度、结晶温度与结晶度均呈降低趋势。这是由于CPL与AH-salt反应制备得到的是无规共聚酰胺6/66, AH-salt的加入,扰乱了晶体结构的完整性,降低了共聚酰胺6/66的熔融温度与结晶度等性质,但并未完全破坏共聚物的晶型。
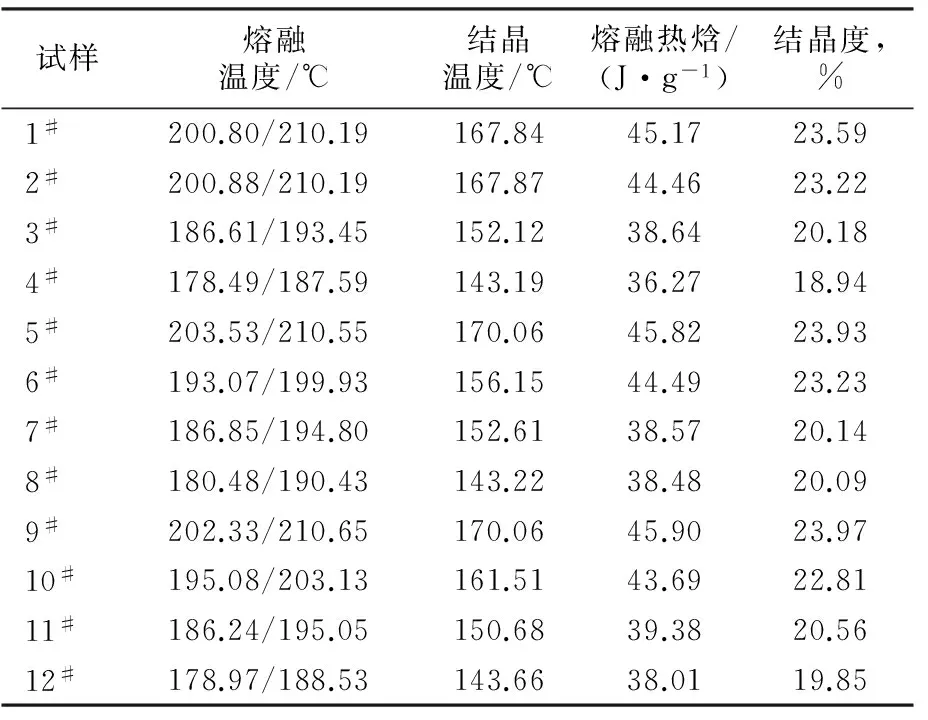
表3 共聚酰胺6/66试样的DSC数据Tab.3 DSC data of copolyamide 6/66 samples
3 结论
a. 在常压条件下成功制备了共聚酰胺6/66。共聚酰胺6/66的W随着AH-salt添加量的增加而降低,开环剂ε-氨基己酸的加入对共聚反应的影响非常明显,W可降低至5.47%。
b. 在高温聚合条件下,当AH-salt质量分数达到20%时, AH-salt易分解并导致配比变化,反应体系中多余的己二酸起到了链终止剂的作用,共聚酰胺6/66的ηr迅速下降。
c.FTIR和XRD分析结果表明,在该实验条件下制备的共聚酰胺6/66均形成了稳定α晶型。
d.DSC分析结果表明,随着共聚组分AH-salt添加量增加,共聚酰胺6/66熔融温度、结晶温度和结晶度均呈减小趋势。同时AH-salt的加入导致了共聚物晶体结构重组,出现了熔融双峰。
[1] 福本修.聚酰胺树脂手册[M].施祖培, 杨维榕, 唐立春,译. 北京:中国石化出版社, 1994: 375-406.
FukumotoOsamu.Polyamideresinhandbook[M].ShiZupei,YangWeirong,TangLichun,trans.Beijing:ChinaPetrochemicalPress, 1994: 375-406.
[2] 何建辉,熊华富,李湘平,等.低熔点聚酰胺的合成与应用研究进展[J].合成纤维工业,2017,40(1): 54-59.
HeJianhui,XiongHuafu,LiXiangping,etal.Researchprogressinsynthesisandapplicationoflow-meltingpointpolyamide[J].ChinSynFiberInd,2017, 40(1): 54-59.
[3]ZhangHeping,ValentineC.Toughenedmonofilaments:US, 11627082[P]. 2007-01-25.
[4]HarveyED,HybartFJ.Ratesofcrystallizationofcopolyamides.II.Randomcopolymersofnylons66and6[J].JApplPolymSci, 1970, 14(8): 2133-2143.
[5]SuehiroK,EgashiraT,ImamuraK,etal.Structuralstudieson6-66and6-68copolyamides[J].Actapolymerica, 1989, 40(1): 4-8.
[6]VasanthanN,SalemDR.FTIRspectroscopiccharacterizationofstructuralchangesinpolyamide-6fibersduringannealinganddrawing[J].JPolymSciPartB:PolymPhys, 2001, 39(5): 536-547.
[7]OkadaA,KawasumiM,TajimaI,etal.AsolidstateNMRstudyoncrystallineformsofnylon6[J].JApplPolymSci, 1989, 37(5): 1363-1371.
[8]HuangYingshan,JiangFeng,LiJie,etal.Controllableformationoftheαandγcrystallinephasesofnylon6[J].JNaturSciHunanNormUniv, 2016, 39(1): 35-42.
[9]RobertsRC.Themeltingbehaviorofbulkcrystallizedpolymers[J].JPolymSciPartBPolymLett, 1970, 8(8):381-384.
[10]FuShuren,ChenTaoyung.Multiplemeltingnylon1010[J].ChinJPolymSci, 1983, 1(2):47-53.
Synthesis and characterization of copolyamide 6/66
Luo Yuhang1,2, Peng Lu1, Jiang Feng2, Zhang Wei1, Yi Chunwang1,3
(1.KeyLaboratoryofHunanHigherEducationforSustainableResourcesProcessingandAdvancedMaterials,HunanNormalUniversity,Changsha410081; 2.StateKeyLaboratoryofBiobasedFiberManufacturingTechnology,ChinaTextileAcademy,Beijing100025; 3.NationalandLocalJointEngineeringLaboratoryforNewPetro-ChemicalMaterialsandFineUtilizationofResources,HunanNormalUniversity,Changsha410081)
A series of polyamide 6/66 copolymers were synthesized by using caprolactam and hexamethylene adipamide as raw materials. The effects of the monomer proportion, ring opening agent and relative molecular mass regulator on the copolymers properties were studied. The structure and thermal properties of the copolymers were characterized by Fourier transform infrared spectrometry,1H nuclear magnetic resonance, X-ray diffraction, differential scanning calorimetry. The results showed that hexamethylene adipamide andε-aminocaproic acid could not only promote the copolymerization reaction, but also decrease the extractable content of copolymer; the addition of hexamethylene adipamide did not change the crystal structure of polyamide 6 with stableαcrystal form; and the double melting peaks of copolyamide 6/66 were due to the reorganization of crystal structure.
polycaprolactam; polyhexamethylene adipamide; copolyamide; synthesis; crystal structure; structural characterization
2017- 02-13; 修改稿收到日期:2017- 04-12。
罗玉航(1991—),男,硕士研究生,主要从事PA 6聚合工程、 功能性聚酰胺的研究。E-mail:lyhsilenthill@163.com。
十三五国家重点研发计划项目(2016YFB0302702)。
TQ316.4+2
A
1001- 0041(2017)03- 0031- 04
* 通讯联系人。E-mail:cwyi@hunnu.edu.cn。
猜你喜欢
青岛科技大学学报(自然科学版)(2023年6期)2023-11-25 17:17:56
现代塑料加工应用(2021年5期)2021-02-28 08:18:26
陶瓷学报(2020年2期)2020-10-27 02:16:14
纺织科学研究(2017年4期)2017-05-17 04:00:03
化工管理(2017年24期)2017-03-06 06:38:40
化工管理(2017年5期)2017-03-05 08:28:57
中国塑料(2016年1期)2016-05-17 06:13:12
中国塑料(2016年5期)2016-04-16 05:25:36
中国塑料(2015年6期)2015-11-13 03:02:34
中国塑料(2015年12期)2015-10-16 00:57:21
