
巴西橡胶树的基因组序列框架图
2017-06-19 15:50田郎
陕西林业科技 2017年2期
田 郎 译
(中国热带农业科学院橡胶研究所,海南 儋州 571737)
巴西橡胶树的基因组序列框架图
田 郎 译
(中国热带农业科学院橡胶研究所,海南 儋州 571737)
巴西橡胶树(Heveabrasiliensis)为大戟科多年生木本植物,也是迄今为止全世界商品天然橡胶(NR)的主要来源。NR是一种具有高弹性、高可塑性及高回弹力的胶乳聚合物,自1876年以来, NR在世界经济发展中一直都起着至关重要的作用。本文报道巴西橡胶树的基因组序列框架图。已组装的基因组序列大约1.1 Gb,约占整个单倍体基因组估计长度(2.15 Gb)的51%。该基因组中大约有78%为重复DNA。通过基因预测共鉴定出68955个基因模型,其中12.7%为橡胶树所特有。此外,大多数与橡胶生物合成、橡胶木材形成、病害抗性以及致敏性有关的关键基因也被成功鉴定。从该基因组获得的序列信息将有助于人们进一步培育高产无性系,以充分满足全世界对天然橡胶不断增长的需求。
巴西橡胶树;大戟科;天然橡胶;基因组
1 背景
橡胶是生产和制造5 000多种产品不可或缺的重要材料[1]。全世界大约有2 500种植物均能合成橡胶[2],但其中只有巴西橡胶树(Heveabrasiliensis)是商品天然橡胶(NR)的主要来源。该大戟科物种起源于南美亚马孙盆地,但直到19世纪才开始大规模用于商业开发,不过其人工驯化栽培并非始于其南美起源地。如今,人工栽培的橡胶树主要分布于亚洲、非洲及拉丁美洲的热带地区。橡胶树大约种植7 a后可开始收获胶乳,其经济寿命约25~30 a。根据国际橡胶研究组织(www.rubberstudy.com)统计, 2011年全球天然橡胶的产量将近1100万t,其中亚洲约占93%。在20世纪,全世界橡胶(包括天然和合成橡胶)的需求量一直呈现稳步上升的态势,预计今后还会继续增长。
NR是一种具有弹性强、可塑性高、回弹力强、 抗冲击力强以及散热性良好等诸多性能的胶乳聚合物[2]。这些优良性能使得NR在许多产品的制造上难以被合成橡胶所取代,如医用手套及飞机和卡车的重型轮胎。NR由94%的顺式-1,4-聚异戊二烯和6%的蛋白质及脂肪酸组成[3]。顺式-1,4-聚异戊二烯这种生物高聚物由含5个碳的异戊烯焦磷酸(IPP)单体构成,并在橡胶粒子的表面通过缩合反应而生成。橡胶链的延伸由顺式-异戊烯基转移酶(CPTs),也称橡胶多聚酶催化[4],所产生的聚合物的分子量是决定橡胶质量的重要因素。仅有为数不多的植物能够大量合成高质量NR(分子量>100万Da),其中包括巴西橡胶树以及潜在的橡胶树替代作物银胶菊(Partheniumargentatum) 和俗称橡胶草的俄罗斯蒲公英(Taraxacumkoksaghyz)[5]。
除了NR之外,当橡胶树进入胶乳生产末期而失去生产价值时,它们还可作为木材加以利用。如今,橡胶木已成为东南亚的一种主要出口木材[1]。橡胶木材的天然色泽及优良物理性质使其非常适于制作实木地板及家具。有鉴于此,人们业已培育出了多个胶木兼优的橡胶无性系。
橡胶树的病害及橡胶制品的致敏性也是橡胶业关注和担忧的重要问题。真菌病害,例如由病原菌Microcyclusulei引起的南美叶疫病(SALB),以及分别由病原菌Colletotrichum、Oidium和Corynespora引起的落叶病是橡胶生产的主要威胁[1]。在20世纪30年代中期,SALB摧毁了整个巴西的植胶业。亚洲的橡胶园虽然尚未受到该病害的侵袭,但此病一旦在该地区爆发将会产生毁灭性的后果。因直接接触某些天然橡胶制品(如橡胶手套)而引发的人体过敏反应一直是医学上关注的重要问题之一。引起这类过敏症的过敏源主要为橡胶树的胶乳中存在的某些蛋白质。近年来,人们已开始尝试将银胶菊作为低致敏性胶乳的生产来源[2]。
常规育种方法的局限性及基因组信息的匮乏和不足已严重地阻碍和制约巴西橡胶树的遗传改良。分子标记辅助选择通过对目标基因型的直接选择可有效地提高育种效率。标记间的遗传连锁分析以及理想表型的遗传定位将进一步提高选择的准确度。近年来,高通量测序技术的迅猛发展及应用[6-12]在一定程度上拓宽并丰富了巴西橡胶树的基因资源。然而迄今为止,该物种的全基因组信息依然十分欠缺。之前人们的研究大都集中于转录组分析,但事实上,基因组的非编码区域对了解控制基因表达的调控元件以及建立一整套更加全面的分子标记必不可少。本文首次报道巴西橡胶树的基因组序列框架图,以期为该重要经济作物的进一步改良提供一个良好的平台。
2 结果与讨论
2.1 基因组测序及注释
本研究对马来西亚橡胶研究院培育的巴西橡胶树高产无性系RRIM600进行了全基因组测序,该无性系的亲本组合为Tjir1×PB86。巴西橡胶树的基因组序列分布于18对染色体上[13],通过孚耳根微显影[14]其单倍体基因组估计约2.15 Gb。我们采用全基因组鸟枪法(whole-genome shotgun,WGS)并利用Roche/454, Illumina, 以及SOLiD测序平台获得大约43×覆盖率的序列数据(表1)。由于大多数测序数据来自Roche/454平台, reads(读取片段)相对较长,尤其是鸟枪片段(shotgun)为单端测序,故我们选用Newbler软件[15]进行最后的组装[16]。对于初步组装的序列我们还对其进行了重复基序的鉴定,并去除与重复区域匹配的原始测序reads,同时整个拼装过程均采用严格的组装参数。在滤除重复匹配reads之后,我们仅根据27.86Gb即大约13×覆盖率的测序数据(表1)最终组装获得总长为1119Mb的序列支架(scaffolds),其N50 scaffold长度为2972bp(表2)。之后,根据154个微卫星标记[17],我们将143个scaffolds以及1325个关联基因锚定到了巴西橡胶树的18个连锁群上。在已定位的scaffolds中,还鉴定出另外74个已报道过的标记。这些标记大部分被定位在基因间隔区。
本研究基于全基因组鸟枪法(WGS)策略,并完全采用新一代测序技术成功构建巴西橡胶树的全基因组框架图,而其它植物中仅有少数几种采用类似方法获得全基因组序列图谱。与我们的研究方法一样,Shulaev V等人在草莓中也是结合采用Roche/454,Illumina以及SOLiD平台的reads(39×覆盖率)构建其全基因组图谱,不过草莓基因组(240Mb)几乎比橡胶树小9倍,而且重复DNA的比例要小得多(22%),在此情况下所得到的contigs/scaffolds相对要大得多[18]。然而,本研究中我们所拼装得到的最大scaffold(531.5 kb)与大麻基因组利用Illumina和Roche/454测序数据组装获得的最大scaffold的长度(565.9 kb)几乎相当[19]。橡胶基因组组装的主要挑战在于其大量重复序列的存在,这也是KFX Mayer等人在大麦基因组(5.1Gb,84%重复DNA)组装过程中所遇到的一个难题,他们在全基因组鸟枪法测序中对Illumina平台产生的短reads进行拼接仅获得长度相对较短的contigs(N50=1425 bp)[20]。不过,当将其与BAC物理图谱及高分辨率遗传图谱相结合时,依然可实现染色体水平上的序列重建。因此,对橡胶树而言,下一步要做的就是结合物理图谱或其它能够提供更多序列毗连信息的方法以进一步改进其基因组拼装的质量。
利用基因组重复序列分析软件RepeatModeler及RepeatMasker,在我们组装的序列中有72.01%被鉴定为重复DNA(不包括低复杂度区域及RNA基因),由此估计重复序列约占橡胶树基因组的78%,这与玉米(85%)[21]和大麦(84%)[20]相当。分析还显示,长末端重复序列(LTR)类反转录转座子为最主要的一类转座元件(占总重复序列的46.15%),其中又以Gypsy类(38.20%)和Copia类(7.38%)为最多。总的重复元件中仅有不到2%为DNA转座子。重复元件中的大部分(50.24%)与任何已知的家族均无关。
注:* 来自Illumina测序平台的 reads 通过去除与重复区域匹配的reads进行过滤,来自SOLiD测序平台的 reads通过只保留长度至少为50 pb的双末端测序reads进行过滤。
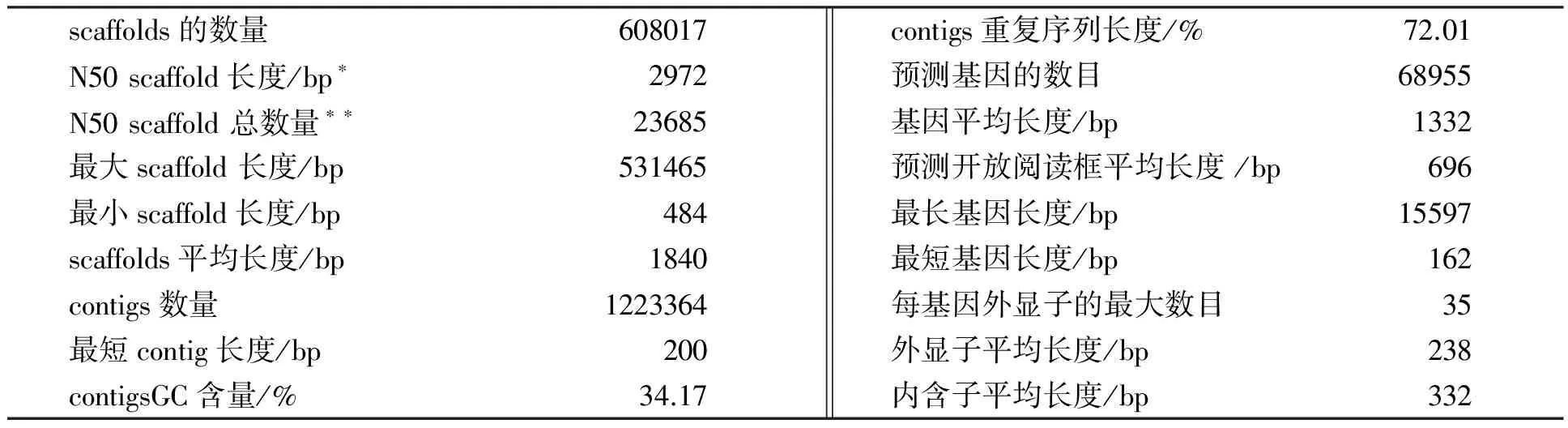
表2 巴西橡胶树基因组组装及注释统计资料
注:*指将所有scaffolds按长度从长到短排序并累加,当累加长度达到所有scaffolds总长度50%时最后1个scaffold的长度;**指将所有scaffolds按长度从长到短排序并累加,当累加长度达到所有scaffolds总长度50%时scaffolds的总数。
结合来自多个从头基因预测程序以及转录组和蛋白质比对的证据和信息,我们采用EVidenceModeler(EVM)软件[22]从已屏蔽的组装序列中预测出68 955个基因模型,基因、外显子及内含子的平均长度分别为1 332 bp、238 bp及332 bp(表2)。在已公布的137 540个橡胶树表达序列标签(ESTs)及组装的转录本中,有95.4%存在于该基因组中。为了给基因模型预测及确认提供进一步的支持,我们还进行了叶片转录组测序,并利用Roche/454及Illumina测序平台分别获得1 085 Mb和4.89 Gb的转录组序列。之后这些序列被从头组装进73 060个contigs(重叠群)。分析显示,这些contigs中99%以上均与组装的基因组序列相匹配,同时,来自Roche/454测序平台的转录组读取片段中有81%也与之相匹配。这些结果表明,我们组装的基因组框架序列中已包含了该植物的大部分基因。
本研究借助不同数据库对来自预测基因的蛋白质序列作了进一步的注释,这些数据库包括NCBI非冗余数据库,SwissPro蛋白序列数据库[23], InterPro 蛋白质家族数据库[24], 以及KEGG生物学通路数据库[25]。真核直系同源蛋白簇(KOG)[26]分析显示,蛋白质的数量在“信号转导机制”( 5 216个),“翻译后修饰, 蛋白质周转, 分子伴侣”(2 886个),以及“碳水化合物转运与代谢”(1 665个)等功能范畴上明显占更高比例。此外, 富含亮氨酸重复(LRR)是该基因组中最为丰富的 Pfam[27]结构域。在预测的基因中,有6.7%含有信号肽,其中大多数被定位在质体及细胞外结构中。
除了蛋白质编码基因之外,我们还鉴定出了729个tRNA基因,其中包括12个抑制(Sup)tRNA,32个假基因,以及4个功能未定的tRNA基因。令人感兴趣的是,我们还观察到了tRNA基因成簇分布的现象,例如所有Sup tRNA基因仅出现在2个scaffolds之中(9个在scaffold 134 351中,3个在scaffold 134 362中)。另外,我们在组装的序列中还鉴定出了5S(113个拷贝)、5.8S(18个拷贝)、18S(11个拷贝)以及28S (21个拷贝) rRNA基因。
2.2 系统发生分析及种系特异性基因
利用来自17个已测序植物基因组的114个单拷贝直系同源基因簇进行系统发育分析,结果显示,巴西橡胶树与木薯(Manihotesculenta)共有一个最近的祖先(图1),这与该植物叶绿体基因组的鉴定分析结果也相一致[28]。除了大戟科之外,与之关系最为密切的已测序基因组为毛果杨(Populustrichocarpa)。与Shulaev V等人基于154个核基因所作被子植物系统发育分析的结果一致[18],我们的分析结果也显示,金虎尾目(包括杨柳科的杨属及大戟科)与锦葵类植物的其它成员拥有一个共同的祖先。
对13个有代表性的植物基因组(分为大戟科、双子叶植物、单子叶植物及低等植物4组)进行比较分析,结果发现,该4组基因组共有一套包括7140个基因家族的核心基因,而有9516个基因家族则为大戟科组所特有(图2a)。比较4个已测序的大戟科植物基因组(即麻风树、蓖麻、木薯和巴西橡胶树),其结果显示,共包含8748个基因的2708个基因家族为橡胶树所特有(图2b)。这些橡胶树特有基因可被归入526个GO[29]功能范畴,266个InterPro 结构域,以及267个Pfam结构域。最为丰富的InterPro 及Pfam结构域是富含亮氨酸重复序列(LRR)及蛋白激酶。KOG分析显示,这些基因中的大多数与信号转导,细胞骨架,以及翻译后修饰有关。
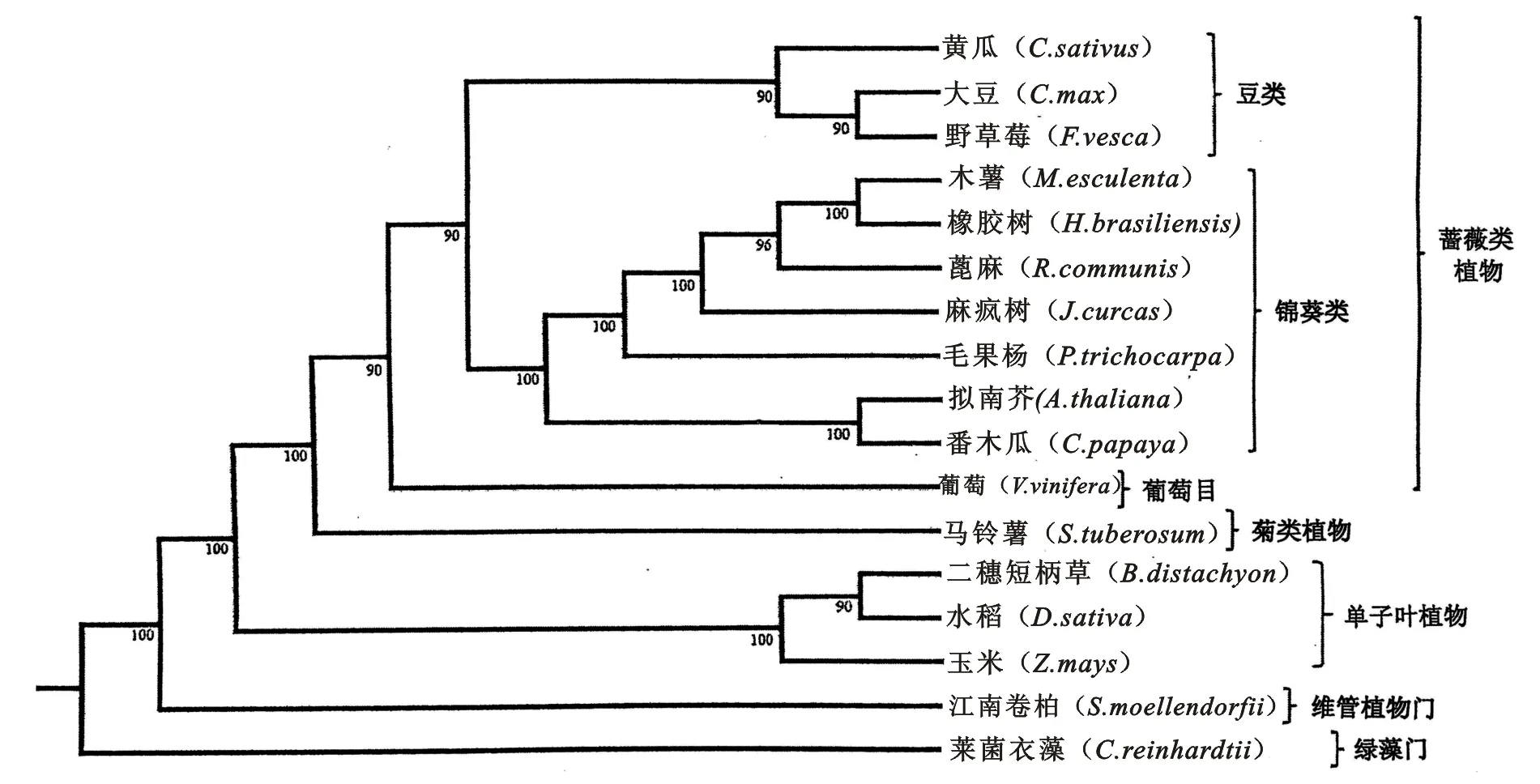
采用PhyML软件并利用分布在17个已测序物种的144个单拷贝直系同源基因簇构建基于最大似然法的系统发生树。最优树的寻找选用SPR(子树修剪与嫁接)和NNI(最近邻居互换)算法中最佳的一个。分析显示巴西橡胶树在分类学上属于锦葵类植物,并与木薯(M.esculenta)的亲缘关系更近。 采用自举法(bootstraping) 检验系统树的可信度(随机重复次数=100,随机种子数目=7),自举检验值(bootstraping值)示于图中每个分支的节点处。
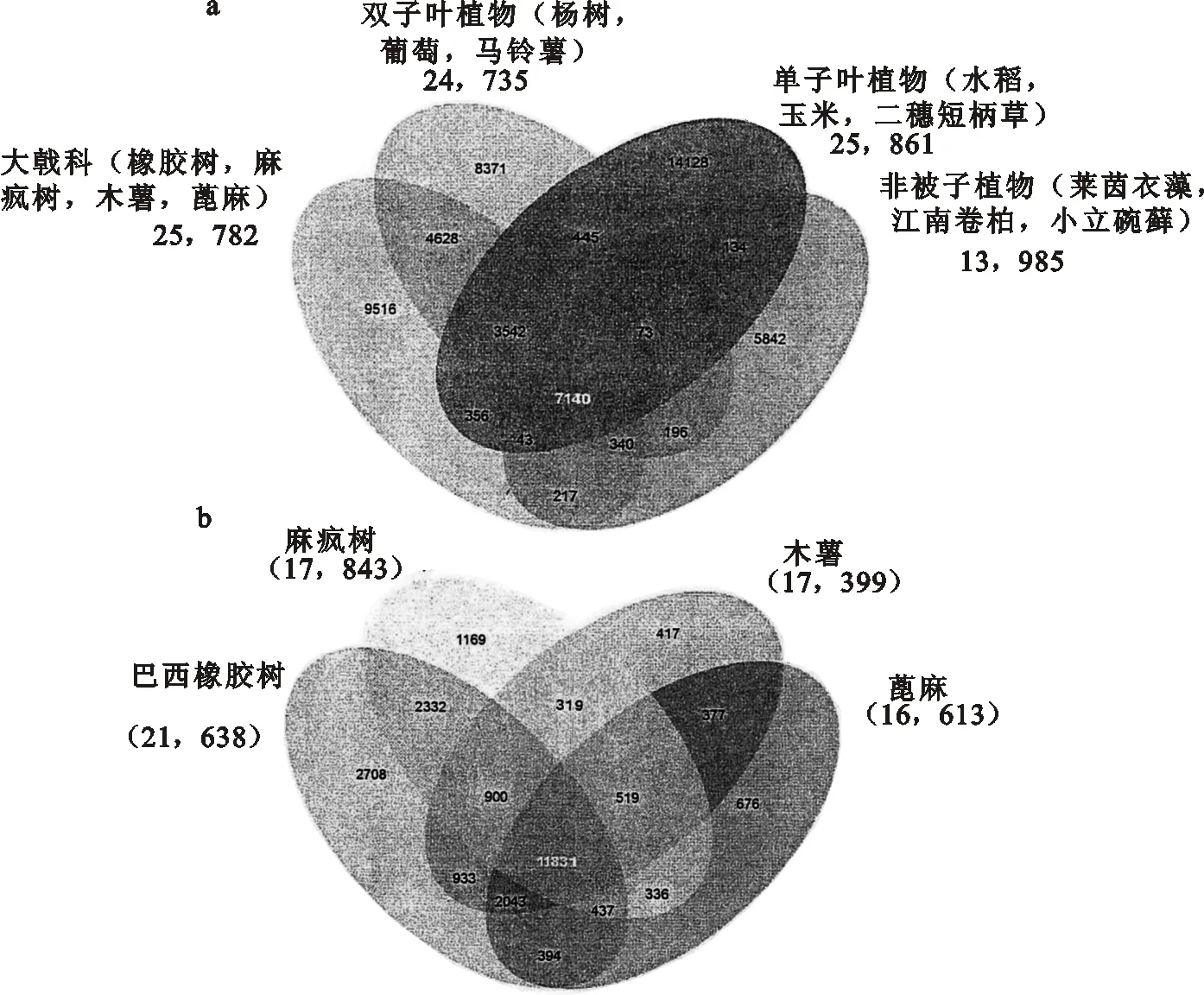
a.利用OrthoMCL软件鉴定出13个植物物种特有及共有的基因家族;b. 利用OrthoMCL软件鉴定出4个已测序大戟科物种特有及共有的基因家族
2.3 橡胶生物合成
橡胶生物合成涉及碳固定、同化物装载运输、橡胶前体物质合成与代谢以及乳管中聚异戊二烯的储存等诸多过程。蔗糖既为橡胶生物合成提供碳骨架又为合成提供能量,而乳管则是其强大的吸收汇[30]。本研究中,我们重构了巴西橡胶树橡胶生物合成的整个代谢途径。橡胶的生物合成过程包括12个主要步骤(图3)并涉及417个基因。我们还确证了这些来自叶片和(或)胶乳cDNA库的基因的表达,并且在大多数基因家族中至少检测到了1个异构体。此外,我们将巴西橡胶树的基因组与得自银胶菊产胶树皮组织的ESTs[31]进行了比较,结果发现,有360个橡胶树的基因与银胶菊ESTs相匹配,其中有205个基因与最佳匹配物的序列一致性高于70%。
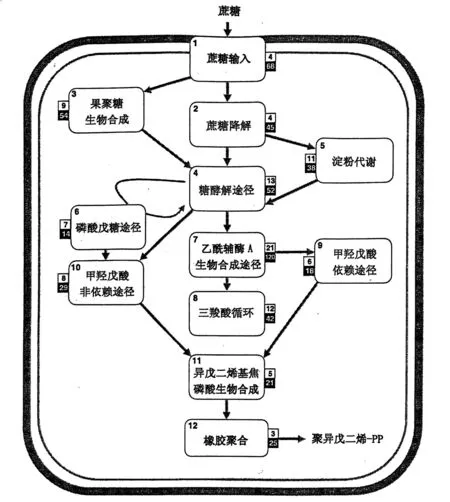
注:白色小框中数字为每个步骤所涉及酶与蛋白的数量,黑色小框中数字为橡胶树中直系同源体的数量。图3 天然橡胶生物合成的12个主要途径示意图
蔗糖转运蛋白及其表达模式与割胶和橡胶生产有直接关系[32]。蔗糖及单糖借助蔗糖和单糖转运蛋白(SUT和MT)被输送进乳管细胞溶质,该2种蛋白质分别由7个和30个基因编码。β-呋喃果糖苷酶和果聚糖β-果糖甙酶能将蔗糖转变成单糖,并且在巴西橡胶树中发现大量此类基因(31个),这充分表明该功能在橡胶生物合成中的重要性。过多的蔗糖则以果聚糖和淀粉形式被储存,并能够用作以后橡胶生物合成的碳源。果聚糖代谢包括9个酶促步骤(由54个基因编码),而淀粉代谢则涉及11个酶促反应(由38个基因编码)。碳通过醣酵解(由52个基因编码)、可供选择的磷酸戊糖途径(由14个基因编码)以及乙酰辅酶A生物合成途径(由120个基因编码)定向生成中间底物,用于橡胶前体物的生物合成。
用于橡胶生物合成的类异戊二烯前体由非自主型的细胞质甲羟戊酸途径(MVA)以异戊烯基焦磷酸(IPP)的形式提供[33]。有人认为自主型的质体甲羟戊酸途径(MEP)也能为橡胶生物合成提供IPP[34]。最近,橡胶树实生苗的13C-标记研究显示,MEP途径为类胡罗卜素的生物合成提供IPP,但并不为橡胶生物合成提供IPP[35]。不过,MVA和MEP途径基因的表达分析显示,在并不产生大量类胡罗卜素的成龄橡胶树或无性系中,MEP途径很可能是提供IPP的另一条替代途径[8]。在巴西橡胶树基因组中,我们鉴定出了18个和29个分别参与MVA及MEP途径的酶基因。活化的烯丙基焦磷酸(法尼基焦磷酸, 香叶基香叶基焦磷酸,或者十一异戊二烯焦磷酸)为引发橡胶生物合成所必需[36]。这些活化物的产生涉及5个酶促步骤并与组装序列中的21个基因有关。
参与类异戊二烯聚合的橡胶聚合酶属顺式-异戊烯基转移酶(CPTs)家族[37]。我们从橡胶树的基因组中鉴定出了8个CPTs(命名为CPT 1-8),并采用最大似然法和MEGA5.05软件[38]推断其进化历史,根据进化关系将其分成3个类群(图4)。我们发现,第2和第3类群中的巴西橡胶树CPTs与其它植物的CPTs(十一异戊二烯焦磷酸合成酶及脱氢多萜醇焦磷酸合成酶)具有同源性,在这些植物中,CPTs 负责短链C5-异戊二烯(C55-C120)的延伸。由CPT 1-3组成的第1类群为巴西橡胶树所特有,该组成员的作用被证明是催化长链C5-异戊二烯的形成[4]。这些CPTs中,仅第3类群中的CPT 4含有内含子,而且该CPT与其它CPT的同源性最小。小橡胶粒子(SRPP)及橡胶延伸因子(REF)是参与橡胶生物合成的另外2种关键蛋白质[5],在我们组装的序列中分别鉴定出其10个和12个相关基因。
2.4 橡胶木材
成年橡胶树当进入胶乳生产末期后,还可作为一种木材来源用于家具及其它产品的制造。木材质量与几个木质纤维素基因密切相关[39]。本研究中,我们在巴西橡胶树中鉴定出了127个相关基因。与拟南芥和杨树相比,我们在橡胶树中鉴定出了36个纤维素合酶(CesA)编码基因,而前2者中仅分别鉴定出10个[40]和18个[41]这样的基因。本研究在巴西橡胶树中也鉴定出与半纤维素生物合成有关的基因,而且α-L-岩藻糖苷酶的数量要多于α-L-岩藻糖基转移酶,这与杨树相似[42]。木质素是一种由木质素单体组成的杂聚物,它决定着木材的质地和硬度。分析显示,橡胶树基因组中参与木质素单体生物合成的一整套基因与杨树基因组存在最大相似性。与杨树基因组相比,橡胶树中咖啡酸O-甲基转移酶(COMT)和肉桂醇脱氢酶(CAD)的数量明显要少(橡胶树和杨树中的COMT分别为10个和41个,CAD分别为5个和24个)。这可能和杨树木材的硬度[39]与橡胶木材不同有关。本研究还鉴定出了一系列参与木质素单体运输、贮藏、调动以及其最后聚合形成木质素的基因。
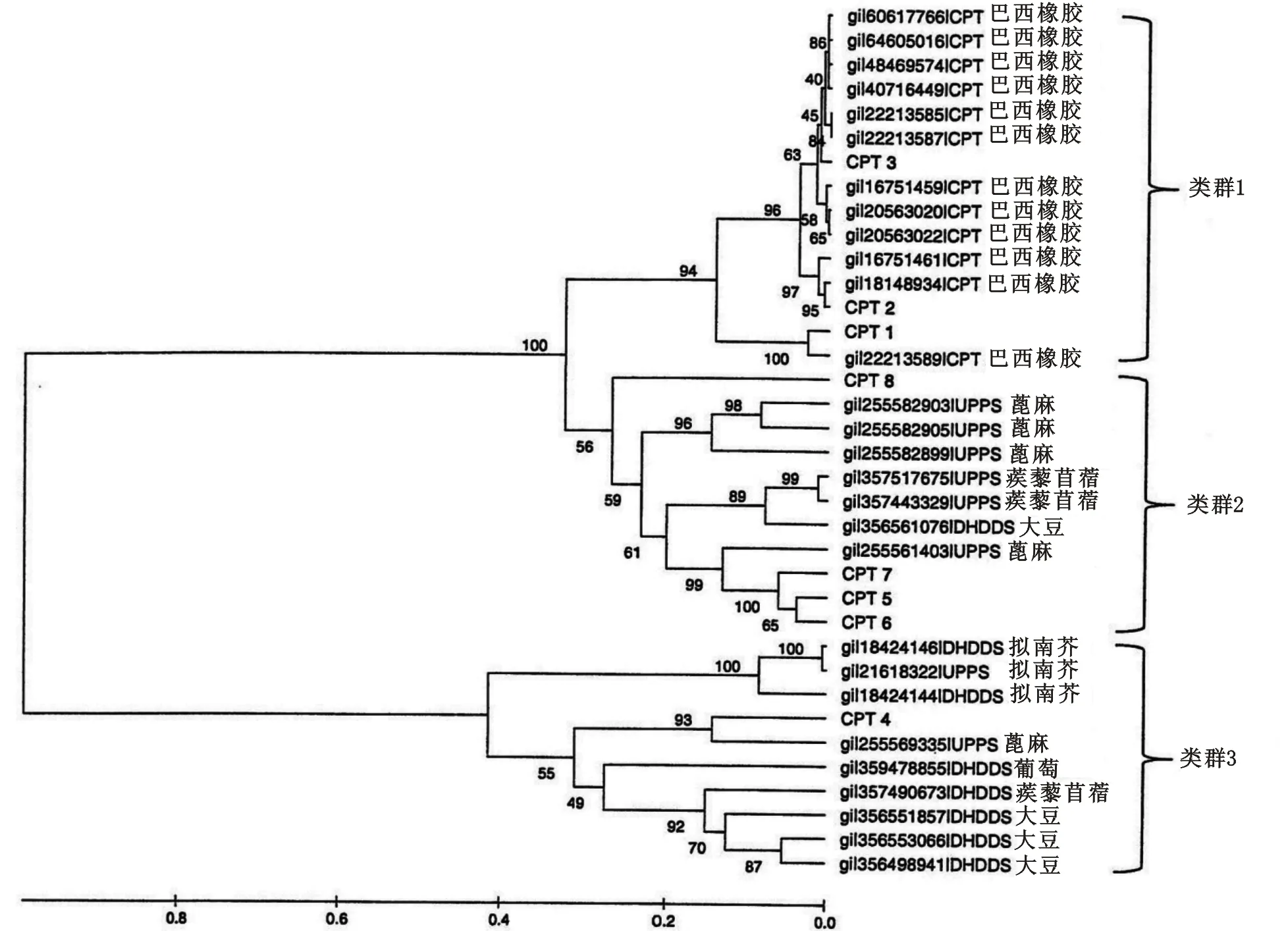
采取最大似然法并利用MEGA5.05软件推断其进化历史。去除所有包含比对空格及缺少数据的位点。CPT,顺式-异戊烯基转移酶;UPPS,十一异戊烯焦磷酸合酶;DHDDS,脱氢多萜醇焦磷酸合酶;自举检验值(重复100次)示于图中分支节点处。
2.5 病害抗性
橡胶树高度感染真菌病害,因此抗病基因的鉴定历来是橡胶树的研究重点之一。超敏反应(HR)是一种能引起植物组织坏死或细胞死亡从而达到限制病原菌生长的早期防御机制。据报道,水杨酸和茉莉酸作为植物信号分子在引发系统获得性抗性(SAR)及诱导某些病程相关蛋白(PR)中起着关键作用[43]。核苷酸结合位点(NBS)编码抗性基因家族在植物中是最大的一类抗性基因[44]。在巴西橡胶树中,我们鉴定出了618个该家族成员,与水稻(Oryza sativa)中鉴定出的数量相当。这些基因被分成6个亚类,即toll-白细胞介素类似受体(TIR)-NBS、卷曲螺旋(CC)-NBS、NBS、TIR-NBS-LRR、CC-NBS-LRR以及NBS-LRR,其中多数不含LRR结构域,而与此相比,其它植物中含有LRR的类型通常更为丰富。我们在组装的基因组中还鉴定出147个病程相关蛋白(PR)和96个早期防御(SAR及HR)相关基因。所有这些病害抗性基因分布在665个序列支架中,并且NBS编码基因通常被发现于这群序列中(如有9个NBS-LRR基因存在于骨架序列409956中)。此外,我们还重构了巴西橡胶树的植物系统获得性抗病性(SAR)及超敏反应(HR)信号途径。所有这些信息均有可能用于对植物生物胁迫的管控。
2.6 胶乳过敏源
天然橡胶胶乳(NRL)过敏症是全球关注的一个重要医学问题。国际公认的NRL过敏源有14种,分别被称为Hevb 1至Hevb 14(www.allergen.org)。在橡胶树中,这些过敏源被100个基因编码。胶乳中的大多数过敏源是含量极为丰富且与胁迫和防御相关的蛋白[45]。导致对天然橡胶胶乳(NRL)过敏的主要过敏源中,Hevb 6 (橡胶素)由16个基因编码,而Hevb 5则是一个单拷贝基因。与橡胶粒子有关的Hevb 1 (REF,橡胶延伸因子)和Hevb 3 (SRPP,小橡胶粒子)分别由12个和10个基因编码。由5个基因编码的Hevb 4 ( 卵磷脂酶同系物)和由9个基因编码的Hevb 13 (酯酶)也被称为含糖过敏源。编码交叉反应蛋白Hevb 8 (抑制蛋白)、Hevb 9( 烯醇酶)以及Hevb 10 (锰超氧化物歧化酶)的基因分别有6个、4个及2个。防御相关过敏源Hevb 2(β-1,3-葡聚糖酶)及Hevb 11 (几丁质酶)各自有11个基因。Hevb 11的结构域分析显示,该结构域存在1个长18~23个氨基酸的信号肽,此信号肽能引发植物的系统伤害反应[46]。除了上述的胶乳过敏源之外,我们还在橡胶树中鉴定出了4类非胶乳过敏源,即花粉过敏源、α-膨胀素、β-膨胀素以及异黄酮还原酶。
2.7 转录因子
巴西橡胶树的基因组大约含有6000个转录因子,它们分布在50个主要家族之中。转录因子约占巴西橡胶树基因模型的8.5%,而其中bHLH、MYB、C3H、G2-like以及WRKY等家族的转录因子所占比例又大大超过其它家族。大多数植物中数量最多的bHLH转录因子家族在橡胶树中多达752个成员。MYB是一个多样化的转录因子家族,该家族的转录因子与bHLH家族转录因子互作从而调节次生代谢[47]及生物和非生物胁迫,在橡胶树中该家族成员的数量也多达570个。C3H家族转录因子涉及花发育、胚胎发生、越冬以及叶片衰老[48],其成员数为470个。之后为G2-like和WRKY家族的转录因子,它们在橡胶树中的成员数分别为461和445个。据报道该2个转录因子家族分别参与了植物的光合调控[49]及免疫应答[50]。此外,橡胶树中成员数较多的转录因子家族还有MYB-related(397个)、NAC(336个)以及ERF(246个)等。MADS-box基因是一类编码与花器官发育有关的转录因子的同源异型基因。MADS-box基因被分为5类,即Mα、Mβ、Mγ、 Mδ (即 MIKC*) 和MIKCc[51],它们在橡胶树中共有112个成员。在这112个成员中有79个属于II型MADS-box基因(Mδ和MIKCc组),而该类型基因在拟南芥、杨树及水稻中的数量为54~67个。与此相比, I型的 MADS-box基因 (包括Mα、Mβ及Mγ组)在橡胶树中的数量只有33个,而其它3个物种中则存在29~94个。此外, 巴西橡胶树中的MADS-box基因仅有12.5%(112个中的14个)以成簇形式存在,而与此相比,番木瓜的MADS-box基因中则有47% 在基因组中成簇存在[52]。
2.8 植物激素生物合成及信号转导
分析显示, 巴西橡胶树中存在为数众多的植物激素生物合成及信号转导相关基因。被子植物基因组通常都含有较大比例的生长素信号通路相关基因,已鉴定出的基因家族达12个。不过,与其它植物相比,橡胶树中有些生长素基因家族成员明显减少,尤其是生长素上调小RNA(SAUR)及生长素(IAA)阻遏蛋白家族成员的数量显著少于杨树、拟南芥及水稻。赤霉素-20-氧化酶是赤霉素生物合成中的一种关键调节酶,在巴西橡胶树中共鉴定出5个直系同源基因,而蓖麻及水稻中仅存在1个这样的基因。乙烯响应元件结合因子(ERF)在橡胶树中共有246个直系同源基因,该数量大大多于其它植物。ERF转录因子数量的增加可能与橡胶树中特定的乙烯依赖途径有关。12-氧-植物二烯酸还原酶(OPR7)是茉莉酸生物合成途径中的一个关键酶,在橡胶树中存在13个编码基因。一氧化氮合成酶参与一氧化氮的生物合成,而一氧化氮在很多植物抗病防御机制中起着特别重要的作用。在拟南芥、蓖麻、杨树以及水稻中,一氧化氮合成酶基因是一个高度保守的单拷贝基因,但在巴西橡胶树种却有4个拷贝。
2.9 光信号转导及生物钟相关基因
光信号转导通路与生物钟相互联系并对植物的生理活动具有十分重要的影响。光是最重要的环境信号之一,来自环境的光信号作用于植物内在的计时机制,即生物钟使植物的生理节奏与环境的昼夜及季节性变化相一致[53]。与杨树和拟南芥相比,巴西橡胶树中涉及光感受及昼夜节律的基因数明显增加,数量多达154个,而杨树和拟南芥中的数量分别仅有77和66个。这些结果表明,环境信号强烈参与橡胶树生理活动及机能的调控。
2.10 F-box蛋白质
F-box蛋白质是SCF(Skp1p-cullin-F-box)蛋白质复合体的组成部分,在植物中SCF复合物广泛参与泛素/26S蛋白酶体途径所介导的蛋白质选择性降解[54]。F-box蛋白质在其N-端存在一个保守F-box结构域(40-50个氨基酸)。据报道,这类蛋白质参与植物众多生长发育过程的调节,如叶片衰老、开花、分枝、光敏素与植物激素信号转导以及自交不亲和等[55]。巴西橡胶树基因组中含有655 个F-box基因,而该家族基因在酿酒葡萄(Vitisvinifera)、番木瓜 (Caricapapaya)、毛果杨、拟南芥 (Arabidopsisthaliana)以及水稻中的数量分别为315、198、425、897以及971个[56]。这一结果颇令人感兴趣,因为与人们的预期正好相反,一年草本生植物的F-box基因家族其基因数量较之木本的多年生植物反而出现了明显的扩张[55]。
2.11 类胡萝卜素
类胡萝卜素在光捕获、光保护、光形态建成、脂质过氧化以及植物的很多发育过程中都起着至关重要的作用[57]。类胡萝卜素在包括橡胶胶乳中的弗莱-威士林粒子(即FW粒子)在内的几乎所有类型质体中都有所发现,而正是FW粒子的存在使得有些橡胶无性系的胶乳呈现出黄色。尽管胶乳中类胡萝卜素的作用尚不十分确定和清楚,但它可能是乳管中异戊烯焦磷酸(IPP)的一个竞争汇。有人认为,在胶乳类胡萝卜素含量低的橡胶无性系中,来自2C-甲基-D-赤藓醇-4-磷酸途径(MEP)的IPP被用于顺式-聚异戊二烯的合成[8]。分析显示,橡胶树中涉及类胡萝卜素生物合成途径的基因数量(48个)较之拟南芥基因组中相关基因的数量(28个)明显增多。八氢番茄红素合成酶和八氢番茄红素脱氢酶是催化类胡萝卜素生物合成中第一个关键步骤的酶,它们在橡胶树中的数量高度增加,基因数分别达5个和9个,而在拟南芥中均为单拷贝基因。这些观察结果表明,橡胶树具有更加有效的类胡萝卜素生物合成机制,而类胡萝卜素在该植物中的合成可能有着多方面的作用和功能。
3 结论
鉴于天然橡胶生产及其可持续性在世界工业及经济发展中的重要作用和地位,橡胶树基因组序列框架图的构建无疑为大戟科植物研究增添了一份宝贵的资料,而这也将有力促进和加速橡胶树分子育种的开展及基因资源的开发利用。我们的观察显示,巴西橡胶树的基因组中存在较大比例的重复序列(大约78%),这有可能缘于该植物非同源重组率及外显子重排事件的增加[58],并由此也降低了后代遗传纯度的一致性。大量重复序列的存在及染色体水平上的信息的缺乏是巴西橡胶树基因组组装的主要障碍。来自全基因组的信息以及与目标基因连锁或相关联的分子标记的鉴定将有助于通过分子育种实现一些重要性状的遗传改良,从而有力地促进天然橡胶生产的发展。与木本植物杨树、桉树及草本模式植物拟南芥相比,全基因组信息的开发利用将特别有助于橡胶树在胶乳产量,木材开发,抗病性,以及降低胶乳致敏性等关键领域取得突破性进展。
4 方法
4.1 基因组测序及组装
从巴西橡胶无性系RRIM600的嫩叶中提取高质量染色体DNA。按试剂盒使用说明书制备鸟枪(Shotgun)及双末端(Paired-end,PE)法测序文库。之后,分别采用Illumina、Roche/454以及SOLiD测序仪产生高质量的读取片段(reads)。Illumina 测序文库包括200 bp及500 bp PE,Roche/454 测序文库包括shotgun、8 kb PE以及20 kb PE, SOLiD 测序文库为2 kb PE。
采用2个为新一代测序数据的从头组装而设计的软件[59],即CLC Workbench (CLC bio, Denmark)和Newbler (version 2.3)进行序列初步组装,该2个组装软件各自采用不同的输入数据内容及组装策略。借助基于de Bruijn图的短序列拼接算法,从经过质量修剪的Illumina 200bp reads生成CLC组装的basic contigs,所有经过修剪的Illumina reads、Roche/454 reads以及SOLiD reads则用于contigs(重叠群)的延伸和连接。Newbler 组装的组装参数设定为:大的或复杂的基因组,reads限定到某一个contig中,最小重叠长度50bp,最小重叠一致性95%。每个组装软件初步组装的序列中,长度≥200bp的Contigs被留下用于进一步分析。
采用RepeatModeler(version 1.0.4) 软件包[60],并按默认参数对2个组装软件初步拼装的序列进行重复序列的从头识别。我们分别从CLC及Newbler软件的初步组装序列中鉴定出2323个和1520个重复模块。之后,通过BLASTX相似性搜索(采用E-值界限10-5),从来自该2个初步组装序列的重复文库中筛选与NR(non-redundant,非冗余)、KEGG以及TrEMBL[61]蛋白质数据库中未分类重复序列相关的潜在基因家族,并将其合并为一个含有3771个重复序列的巴西橡胶树特异性重复文库。以重复序列的出现频率为标准,对该巴西橡胶树重复文库进行筛选,凡是每一个初装序列中出现100次以上的重复序列被保留下来,并将其组合成一个巴西橡胶树高频重复文库,用于软件RepeatMasker (version 3.2.9)[62]的输入。之后将其用于鉴定和屏蔽Newbler初步组装序列中的重复区域,但不包括序列中的低复杂度区域和RNAs。将已屏蔽重复的Newbler初步组装序列作为模板用以筛选通过Illumina平台产生的与重复序列相关的测序reads。在进行筛选之前,Illumina reads已经过质量控制且所有测序碱基位的质量值≥25的reads被保留下来,其中200 bp测序文库的read长度为100 bp,500 bp的双末端测序文库其正向和反向read长度分别为85 bp和75 bp。采用BOWTIE (version 0.12.7)软件将每一个经过质量筛选的高质量Illumina read的前50 bp与Newbler初步组装的序列进行比对,允许的错配碱基不超过3个。与重复区域匹配的成对或非成对reads均从read数据集中排除。就通过SOLiD平台产生的测序reads而言,其质量控制首先是将SOLiD reads与已经SOAP软件[63]校正的Illumina 200 bp reads用较早版本CLC软件初步拼装所得到的序列进行比对,任何种类的匹配错误(包括彩色空隙、单核苷酸差异,或者插入缺失)允许出现2个。两条reads都匹配的SOLiD 成对reads其对应的参考序列被分离出来,并将其作为一个read对。将所有两端读长至少达到50 bp的read对保留于read数据集中。
利用所有Roche/454 reads、部分挑选出的Illumina reads(包括来自200 bp测序文库的成对或非成对reads和来自500 bp测序文库的成对reads)以及SOLiD 成对reads,并采用Newbler组装软件获得最终的基因组组装序列(表1)。从TIGR植物重复序列数据库及TIGR玉米重复序列数据库获得其它植物的重复序列文库,并且核糖体DNA序列已从这些数据库中被去除。将巴西橡胶树特异性重复文库与TIGR植物重复序列数据库合并为RepeatMasker的输入重复文库,以鉴定和屏蔽最终拼装所得contigs中的重复区域,但不包括其所含低复杂度区域及RNAs基因。采用 BOWTIE程序(version 0.12.7)[64]将每一个基因组测序reads的前50 bp比对到最终的基因组拼装序列上,同时利用TopHat (version 1.1.4)软件[65]将通过Roche/454平台产生的转录组测序reads与最终的基因组拼装序列进行比对,以评估编码区域的完整性和覆盖度。
为了鉴定带有细胞器来源的contigs,采用BLASTN搜索程序将拼装序列与巴西橡胶树叶绿体基因组序列进行相似性比对,同时,还通过BLASTX程序将拼装序列与来自豆目植物细胞器基因组,巴西橡胶树、麻风树 (Jatropha curcas)和木薯的叶绿体基因组,以及蓖麻(Ricinuscommunis)、西瓜(Citrulluslanatus)和西葫芦(Cucurbitapepo)线粒体基因组的蛋白质进行比对分析。源于细菌污染的contigs则通过搜索GenBank数据库进行鉴定并将其从最终的拼装序列中去除。
4.2 转录组测序
从巴西橡胶树的叶片中分离总RNA。按试剂盒使用说明书步骤,将分离得到的mRNA随机打断后,通过逆转录合成双链cDNA片段并用其构建测序文库,之后采用Illumina和Roche/454平台测序。利用CLC Workbench组装软件并通过拼接Illumina reads获得最初的转录组序列。将来自Illumina转录组组装的Contigs切割成最大1999 bp的短片段,并与Roche/454转录组测序reads相结合,用于Newbler组装程序的输入用以优化EST数据。最后,通过对非冗余蛋白质数据库进行BLASTX搜索(E-值界限10-5)对转录组组装序列的Contigs进行注释,并以此检测转录组的完整性及多样性。
4.3 基因组注释
利用EVidenceModeler(EVM;version r03062010)软件,通过整合来自转录组比对、蛋白质比对以及基因从头预测的证据最终完成橡胶树已屏蔽基因组的基因模型预测。利用PASA (version r09162010)[66]和Exonerate(version 2.2.0)[67]软件将来自橡胶树转录组组装的Contigs与其基因组序列进行比对。采用GMAP (version 20100727)程序[69]将从PlantGDB数据库[68]获得的植物唯一转录本(PUTs)与基因组进行比对。利用AAT(version 1.52)[70]及BLAT(version 34)[71]软件将从PlantGDB数据库所获基因组测序计划的植物蛋白质序列与组装的橡胶树基因组进行比对。将来自橡胶树转录组组装的Contigs作为训练集,用以训练从头预测软件Fgenesh[72]。之后,将基于转录组比对的PASA预测结果进一步作为训练集并用以训练从头开始的基因预测软件Augustus (version 2.5)[73]、 GlimmerHMM (version 3.0.1)[74]以及SNAP(version 2010-07-28)[75]。此外,在结构注释过程中,我们还一并使用了用拟南芥训练的从头预测软件GeneMarkHMME (version 3)[76]及用甜瓜(Cucumis melo)训练的从头预测软件Geneid (version 1.4.4)[77]进行橡胶树的基因模型预测。最后,利用EVM软件整合来自不同方法及软件的预测结果以获得基因的一致性序列。综合考虑各种预测方法的可信度及有效性等因素,我们以橡胶树转录组比对的PASA预测结果作为最高标准,将各种来源证据的权重人工设置为,橡胶树转录组组装:PASA 1,Exonerate 0.5;植物PUTs:GMAP 0.2;植物蛋白质:AAT 0.2,BLAT 0.2;基因从头预测:Fgenesh 0.6,Augustus 0.5,SNAP 0.3,GlimmerHMM 0.3,Geneid 0.2,GeneMarkHMME 0.2。
为了确保质量和精确的注释,除了常用的功能注释程序外,我们还设置了人工校对及若干验证标准。采用BLASTP程序将来自预测基因的蛋白质序列与数据库Swiss-Prot、TrEMBL、PlantGDB、UniRef100、NCBI非冗余数据库、STRING (version 8.3)[78]以及KEGG GENES进行相似性比对(E-值界限10-5),从而得到相应的功能注释。基于开放阅读框(ORFs)的功能筛查其长度覆盖率及相似性均至少达到70%。对未经完整ORFs得到注释的序列,则进一步通过扫描InterPro、PANTHER[79]、PRINTS[80]、PROSITE[81]patterns、Pfam以及SMART[82]等数据库进行结构域检测,随后与已经得到明确注释的序列模板(如拟南芥和蓖麻)进行比对得到相关功能信息。之后,基于相互最佳匹配法并借助Pathway Studio (Ariadne Genomics Inc.)软件从严格审核的植物特定数据库中寻找直系同源物,并以此对功能注释作进一步的分类。利用Pathway Studio功能类别及KEGG直系同源评估获得酶的EC分类号。根据STRING及GENES数据库的BLASTP匹配片段获得其KO分类号。从InterPro数据库的搜索结果获得蛋白质对应的GO分类号。通过蛋白质与PlantRefSeq、KEGG、Swiss-Prot以及InterPro数据库的比较进行人工审核校对。超过10 000条蛋白质的相应功能得到人工注释。鉴定百分率、序列比对可靠性得分(bit-score)以及长度覆盖率的比较连同其它分析通过计算机的综合处理分析最终使一个特定ORF的推定功能得到可靠认定。
利用TRNAscan-SE v.1.23软件包且EufindRNA检测软件按非严格设置(Int Cutoff=-32.1)鉴定所组装的基因组序列中的tRNA 基因[83]。采用BLASTN 2.2.24软件,通过与拟南芥及水稻所含5S、5.8S、18S及28S rRNA的比对鉴定rRNA基因(长度覆盖率至少为80%,序列一致性至少达到50%[84])。利用SignalP 4.0软件预测和鉴定基因组中的信号肽[85]。
4.4 巴西橡胶树基因家族的鉴定和注释
利用CLC软件并采用合适的模板序列鉴定与橡胶及木质纤维素生物合成、系统获得性抗性、超敏反应、病程相关蛋白、过敏源、转录因子、植物激素代谢及信号转导、生物钟及光信号转导、F-box以及类胡萝卜素生物合成等相关的基因。通过在PlantGDB、UniProtKB/TrEMBL以及Plant_refseq蛋白质数据库进行BLASTX搜索(E-值<10-5)对鉴定的基因作进一步注释。
4.5 NBS-LRR基因家族的鉴定和注释
采用BLASTP程序(E-值<10-5)从PlantGDB、NCBI以及TAIR数据库中搜索与拟南芥NBS-LRR(核苷酸结合位点—富含亮氨酸重复序列)蛋白质相似且覆盖率大于90%的巴西橡胶树蛋白质,并通过与NCBI保守结构域数据库进行结构域匹配加以进一步确认。之后,利用数据库Pfam、InterPro及HMMPanther对这些来自橡胶树基因组的已注释基因模型进行结构域扫描和搜索,并分别获得相应的模体及ID号,其中包括:TIR[PF01582、IPR000157],NBS[PF0931、IPR002182],TIR-NBS [PF01582、PF00931、IPR000157、IPR002182],NBS-LRR[PF0931、PF00560、PF07723、PF07725及PTHR23155:SF236],TIR-NBS-LRR[PF01582、PF0931、PF00560、PF07723、PF07725、IPR000157、IPR002182、IPR001611、IPR011713及PTHR23155:SF300],CC-NBS-LRR[PTHR23155: SF231],以及三种类型的LRR,如LRR_1[PF00560、IPR001611]、LRR_2[PF07723]以及LRR_3[PF07725、IPR011713]。利用COILS 程序预测和鉴定[86]卷曲螺旋(CC)结构域。通过合并分析、人工验证及片段匹配度的检验,我们从巴西橡胶树中鉴定出了618个NBS-LRR基因。
4.6 橡胶生物合成相关基因与银胶菊ESTs的比较
将来自巴西橡胶树与橡胶生物合成相关的基因翻译成蛋白质序列,并用作查询序列与银胶菊ESTs[GenBank:GW775573-GW 787311]作TBLASTN比对分析。所得结果采用E-值界限10-5进行甄别和过滤。
4.7 通路重建
利用Resnet-Plant 3.0数据库及代谢通路数据库(Metabolic Pathway Databases,MPW)[87]并借助Pathway Studio software软件(Ariadne Genomics Inc.)重建巴西橡胶树的代谢通路。Resnet-Plant 3.0数据库集成了从拟南芥AraCyc 4.0数据库引入的303条代谢通路信息,每条代谢通路体现为一系列不同功能类群的蛋白质(酶)及一组相应的化学反应。数据库中的每一个功能类群可包含数量不限并具有相应酶活性编码序列的蛋白质成员。同一功能类群中通常包含若干行使酶活功能所必需的催化及调节亚基的旁系同源物。
利用Pathway Studio初步重建的代谢通路表征为一系列不同功能类群的人工审核蛋白质。该重建过程实际上也相当于一个在代谢网络中填补空缺的过程。带有橡胶基因组标识的蛋白质在Resnet-Plant 3.0数据库中得到注释并去除非橡胶蛋白质之后,有311个功能类群不存在任何与其对应的蛋白质成员。利用根据BLASTP结果采用双向最佳匹配法鉴定出的直系同源蛋白质,并通过对已组装橡胶基因组DNA序列的TBLASTN分析人工鉴定自动注释未能发现的蛋白质。填补橡胶代谢网络中的空缺首先是从GenBank及UniProt数据库下载能执行所缺失酶蛋白功能的蛋白质序列,然后将其作为查询序列用于TBLASTN分析。这些用于查询的蛋白质序列或者来自拟南芥基因组,或者是来自其它植物或细菌基因组。通过与拟南芥AraCyc数据库的比较,鉴定仅仅存在于水稻RiceCyc或杨树PoplarCyc数据库中的代谢通路。拟南芥AraCyc数据库中所缺少的代谢通路采用手动方式添加到Pathway Studio的巴西橡胶数据库中。MPW数据库也被用于橡胶生物合成途径的重建。
4.8 将序列支架锚定到连锁群
根据已公布的连锁图谱[17],将组装的scaffolds锚定和定位到橡胶树的18个连锁群。所用154个微卫星标记序列均得自公共序列数据库。通过标记序列与所有scaffolds之间的 BLAST相似性分析对各个scaffolds进行鉴定,而所在scaffold上的基因模型也因此得以鉴定并被锚定在连锁群的相应位置。当某个scaffold上存在一个以上标记时,基因则有可能被锚定在正确的位置上,也有可能在其它位置,其定位不确定。位于scaffolds上的其它额外标记则通过全部scaffolds对GenBank数据库的BLAST相似性分析进行鉴定。
4.9 特有及共有基因簇分析
采用OrthoMCL软件[88]鉴定和估算大戟科植物以及不同植物类群间旁系和直系同源基因簇的数量。采用标准设置(BLASTP, E-值<10-5)计算序列的多对多相似性。
4.10 系统发生分析
采用17个已测序的植物基因组,即莱茵衣藻(Chlamydomonasreinhardtii)、江南卷柏(Selaginellamoellendorfii)、玉米(Zeamays)、水稻、二穗短柄草(Brachypodiumdistachyon)、马铃薯(Solanumtuberosum)、葡萄、番木瓜、拟南芥、毛果杨、麻疯树、蓖麻、巴西橡胶树、木薯、野草莓(Fragariavesca)、大豆(Glycinemax)以及黄瓜(Cucumissativus)构建系统发生树。采用BLAST算法及E-值界限10-5对蛋白质序列进行多对多的相似性比较,并从该BLAST分析结果计算鉴定百分率。利用OrthoMCL软件鉴定内旁系同源基因、直系同源基因以及共直系同源基因。通过寻找某一物种内的所有成对蛋白质确定潜在的内旁系同源基因对,这些成对蛋白质之间的序列匹配度高于或等于它们中的任一蛋白质与其它物种中所有蛋白质间的匹配度。通过寻找存在于2个物种间的所有成对蛋白质确定所有潜在的直系同源基因对,这些成对蛋白质之间的序列匹配度等于或高于二者中的任一蛋白质与另一物种中任何其它蛋白质间的匹配度,也即该成对的2个蛋白质互为最佳匹配蛋白质。潜在的共直系同源基因对也通过寻找两个物种间的所有成对蛋白质加以确定,某一物种中的内旁系同源基因通过与另一物种中的内旁系同源基因间的直系同源关系而结为共直系同源基因对。对采用MCL程序对同源基因进行聚类分析,所有存在内旁系及直系同源关系的蛋白质被归入同一类,共产生57 250个同源基因簇。去除不包括所有17个物种的同源基因簇后,共得到1 364个基因簇。通过选留至少在该17种植物的14个成员中均存在单拷贝的同源基因簇对其进一步过滤,最后获得144个单拷贝直系同源基因簇。利用ClustalX软件进行序列比(采用Gonnet系列矩阵,空位开放罚分=10,空位延伸罚分=0.1),之后采用Gblocks软件从比对结果中提取保守序列区。最后,按最大似然法并利用PhyML软件[89]构建系统发生树。采用SPR(子树修剪与嫁接)和NNI(最近邻居互换)算法中最佳的一种寻找最优树。通过自举分析(Bootstrapping)检验系统树的可信度(随机重复100次,随机种子数目为7)。
4.11 基因的实验确认
利用RNeasy Mini Kit (Qiagen)试剂盒并按照使用说明书从巴西橡胶无性系RRIM 600的嫩叶和胶乳中分离总RNA。之后,采用试剂盒SuperScript VILO cDNA Synthesis Kit (Invitrogen)合成cDNA第一链。以cDNA为模板并采用基因特异性引物对基因进行PCR扩增。将纯化的PCR产物克隆到pCR4Blunt-TOPO (Invitrogen)或pJET1.2/blunt (Fermentas)载体中并测序。与橡胶生物合成、木质素生物合成、病害抗性、过敏源、转录因子以及植物激素生物合成相关的基因均按以上步骤扩增、克隆和测序。
4.12 数据的可得性
该全基因组鸟枪法测序计划已提交到DDBJ/EMBL/GenBank数据库,其GenBank登录号:AJJZ00000000。本文所描述的版本为第一版。组装的转录本也已提交到NCBI转录组鸟枪组装数据库,其GenBank登录号:JT914190-JT981478。该橡胶基因组游览器可供下载和使用[90]。
本项目受马来西亚高等教育部顶尖大学计划(APEX)资助,由马来西亚理科大学、中国南开大学泰达生物技术研究院、美国夏威夷大学、美国Ariadne Genomics公司、丹麦CLC bio公司、南非德班理工大学、美国德克萨斯A&M大学等机构共同完成。Ahmad Yamin Abdul Radman等所有署名作者均已阅读并同意本论文的最终稿。
[1] Prabhakaran Nair KP. The Agronomy and Economy of Important Tree Crops of the Developing World. Burlington: Elsevier, 2010.
[2] Mooibroek H et al.. Alternative sources of natural rubber. Appl Microbiol Biotechnol 2000, 53:355-365.
[3] Sakdapipanich JT. Structural characterization of natural rubber based on recent evidence from selective enzymatic treatments. J Biosci Bioeng 2007, 103:287-292.
[4] Asawatreratanakul K et al. Molecular cloning, expression and characterization of cDNA encoding cis-prenyltransferases from Hevea brasiliensis: a key factor participating in natural rubber biosynthesis. Eur J Biochem 2003, 270:4671-4680.
[5] Gronover CS et al.. Natural rubber biosynthesis and physic-chemical studies on plant derived latex. In Biotechnology of Biopolymers. Edited by Elnashar M. Croatia: Intech Open Acess Publisher; 2011:75-88.
[6] Triwitayakorn K et al.. Transcriptome sequencing of Hevea brasiliensis for development of microsatellite markers and construction of a genetic linkage map. DNA Res 2011, 18:471-482.
[7] Xia Z et al.. RNA-Seq analysis and de novo transcriptome assembly of Hevea brasiliensis. Plant Mol Biol 2011, 77: 299-308.
[8] Chow KS et al.. Metabolic routes affecting rubber biosynthesis in Hevea brasiliensis latex. J Exp Bot 2012, 63:1863-1871.
[9] Gebelin V et al.. Identification of novel microRNAs in Hevea brasiliensis and computational prediction of their targets. BMC Plant Biol 2012, 12:18.
[10] Li D et al.. De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genomics 2012, 13:192.
[11] Lertpanyasampatha M et al.. Genome-wide analysis of microRNAs in rubber tree (Hevea brasiliensis L.) using high-throughput sequencing. Planta 2012, 236:437-445.
[12] Pootakham W et al.. Development of genomic-derived simple sequence repeat markers in Hevea brasiliensis from 454 genome shotgun sequences. Plant Breeding 2012, 131:555-562.
[13] Leitch AR et al.. Molecular cytogenetic studies in rubber, Hevea brasiliensis Muell. Arg. (Euphorbiaceae). Genome 1998, 41: 464-467.
[14] Bennett MD et al.. Nuclear DNA amounts in angiosperms-583 new estimates. Ann Bot 1997, 80:169-196.
[15] Margulies M et al.. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437:376-380
[16] Barthelson R et al.. Plantagora: modeling whole genome sequencing and assembly of plant genomes. PLoS One 2011, 6: e28436.
[17] Le Guen V et al.. A rubber tree’s durable resistance to Microcyclus ulei is conferred by a qualitative gene and a major quantitative resistance factor. Tree Genet Genomes 2011, 7: 877-889.
[18] Shulaev V et al.. The genome of woodland strawberry (Fragaria vesca). Nat Genet 2011, 43:109-116.
[19] van Bakel H et al.. The draft genome and transcriptome of Cannabis sativa. Genome Biol 2011, 12:R102.
[20] The International Barley Genome Sequencing Consortium. A physical, genetic and functional sequence assembly of the barley genome. Nature 2012, 491:711-716.
[21] Schnable PS et al.. The B73 maize genome: complexity, diversity, and dynamics. Science 2009, 326:1112-1115.
[22] Haas BJ et al.. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol 2008, 9:R7.
[23] Boutet E et al.. UniProtKB/Swiss-Prot. Methods Mol Biol. 2007,406:89-112.
[24] Zdobnov EM et al.. InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics 2001, 17:847-848.
[25] Kanehisa M et al.. The KEGG resource for deciphering the genome. Nucleic Acids Res 2004, 32:D277-D280.
[26] Tatusov RL et al.. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 2003, 4:41.
[27] Finn RD et al.. Pfam: clans, web tools and services. Nucleic Acids Res 2006, 34:D247-D251.
[28] Tangphatsornruang S et al.. Characterization of the complete chloroplast genome of Hevea brasiliensis reveals genome rearrangement, RNA editing sites and phylogenetic relationships. Gene 2011, 475:104-112.
[29] Ashburner M et al.. Gene Ontology: tool for the unification of biology. Nat Genet 2000, 25:25-29.
[30] Silpi U et al.. Carbohydrate reserves as a competing sink: evidence from tapping rubber trees. Tree Physiol 2007, 27: 881-889.
[31] Ponciano G et al.. Transcriptome and gene expression analysis in cold-acclimated guayule (Parthenium argentatum) rubber-producing tissue. Phytochemistry 2012, 79:57-66.
[32] Tang C et al.. The sucrose transporter HbSUT3 plays an active role in sucrose loading to laticifer and rubber productivity in exploited trees of Hevea brasiliensis (para rubber tree). Plant Cell Environ 2010, 33:1708-1720.
[33] Kekwick RGO: The formation of isoprenoids in Hevea latex. In Physiology of Rubber Tree Latex. Edited by d’Auzac J, Jacob JL, Chrestin L. Boca Raton: CRC Press; 1989:145-164.
[34] Ko JH et al.. Transcriptome analysis reveals novel features of the molecular events occurring in the laticifers of Hevea brasiliensis (para rubber tree). Plant Mol Biol 2003, 53: 479 -492.
[35] Sando T et al.. Cloning and characterization of the 2-C-methyl-D-erythritol 4-phosphate (MEP) pathway genes of a natural-rubber producing plant. Hevea brasiliensis. Biosci Biotechnol Biochem 2008,72: 2903-2917.
[36] Rattanapittayaporn A et al.. Significant role of bacterial undecaprenyl diphosphate (C55-UPP) for rubber synthesis by Hevea latex enzymes. Macromol Biosci 2004, 4:1039-1052.
[37] Kharel Y et al.. Molecular analysis of cis-prenyl chain elongating enzymes. Nat Prod Rep 2003, 20:111-118.
[38] Tamura K et al.. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011, 28:2731-2739.
[39] Dillon SK et al.. Allelic variation in cell wall candidate genes affecting solid wood properties in natural populations and land races of Pinus radiata. Genetics 2010, 185:1477-1487.
[40] Richmond TA et al.. The cellulose synthase superfamily. Plant Physiol 2000, 124:495-498.
[41] Djerbi S et al.. The genome sequence of black cottonwood (Populus trichocarpa) reveals 18 conserved cellulose synthase (CesA) genes. Planta 2005, 221: 739-746.
[42] Tuskan GA et al.. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006,313:1596-1604.
[43] Durrant WE, Dong X. Systemic acquired resistance. Annu Rev Phytopathol 2004, 42:185-209.
[44] Mun JH et al.. Genome-wide identification of NBS-encoding resistance genes in Brassica rapa.Mol Genet Genomics 2009, 282:617-631.
[45] Yeang HY et al.. Allergenic protein of natural rubber latex. Methods 2002, 27:32-45.
[46] Ryan CA et al.. Systemic wound signaling in plants: a new perception. Proc Natl Acad Sci USA 2002, 99:6519-6520.
[47] Feller A et al.. Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J 2011, 6:94-116.
[48] Wang D et al.. Genome-wide analysis of CCCH zinc finger family in Arabidopsis and rice.BMC Genomics 2008, 9:44.
[49] Riano-Pachon DM et al.. Green transcription factors: a Chlamydomonas overview. Genetics 2008, 179:31-39.
[50] Rushton PJ et al.. WRKY transcription factors. Trends Plant Sci 2010, 15:247-258.
[51] Arora R et al.. MADS-box gene family in rice: genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics 2007, 8:242.
[52] Ming R et al.. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 2008, 452:991 -996.
[53] Mas P: Circadian clock signaling in Arabidopsis thaliana: from gene expression to physiology and development. Int J Dev Biol 2005, 49:491-500.
[54] Jain M et al.. F-box proteins in rice. Genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiol 2007, 143:1467-1483.
[55] Yang X et al.. The F-box gene family is expanded in herbaceous annual plants relative to woody perennial plants. Plant Physiol 2008, 148:1189-1200.
[56] Hua Z et al.. Phylogenetic comparison of F-box (FBX) gene superfamily within the plant kingdom reveals divergent evolutionary histories indicative of genomic drift. PLoS One 2011, 6:e16219.
[57] Chaudhary N et al.. Carotenoid biosynthesis genes in rice: structural analysis, genome-wide expression profiling and phylogenetic analysis. Mol Genet Genomics 2010, 283:13-33.
[58] Yang S et al.. Repetitive element-mediated recombination as a mechanism for new gene origination in Drosophila. PLoS Genet 2008, 4:e3.
[59] Miller JR et al.. Assembly algorithms for next-generation sequencing data.Genomics 2010, 95:315-327.
[60] RepeatModeler. http://www.repeatmasker.org/RepeatModeler.html webcite
[61] Boeckmann B et al.. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 2003, 31: 365-370.
[62] RepeatMasker. http://www.repeatmasker.org.
[63] Li R et al.. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 2010, 20:265-272.
[64] Langmead B et al.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009, 10:R25.
[65] Trapnell C et al.. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25:1105-1111.
[66] Haas BJ et al.. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 2003, 31: 5654-5666.
[67] Slater GS et al.. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 2005, 6:31.
[68] Dong Q et al.. Comparative plant genomics resources at PlantGDB. Plant Physiol 2005, 139:610-618.
[69] Wu TD et al.. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21: 1859-1875
[70] Huang X et al.. A tool for analyzing and annotating genomic sequences. Genomics 1997, 46:37-45.
[71] Kent WJ: BLAT-the BLAST-like alignment tool. Genome Res 2002,12:656-664.
[72] Salamov AA et al.. Ab initio gene finding in Drosophila genomic DNA. Genome Res 2000, 10:516-522.
[73] Stanke M et al.. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res 2005, 33:W465-W467.
[74] Majoros WH et al.. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20: 2878-2879
[75] Korf I: Gene finding in novel genomes. BMC Bioinformatics 2004,5:59.
[76] Ter-Hovhannisyan V et al.. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res 2008, 18:1979-1990.
[77] Blanco E et al.. Using geneid to identify genes. In Current Protocols in Bioinformatics. Edited by Baxevanis AD, Davison DB. New York: John Wiley and Sons Inc; 2002.Unit 4.3.
[78] Szklarczyk D et al.. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 2011, 39:D561-D568.
[79] Thomas PD et al.. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res 2003, 31:334-341.
[80] Attwood TK et al.. The PRINTS protein fingerprint database: functional and evolutionary applications. In Encyclopaedia of Genetics, Genomics, Proteomics and Bioinformatics. Edited by Dunn M, Jorde L, Little P, Subramaniam A. New Jersey: John Wiley and Sons Ltd; 2006.
[81] Sigrist CJA et al.. PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res 2010, 38:D161-D166.
[82] Letunic I et al.. SMART 7. recent updates to the protein domain annotation resource. Nucleic Acids Res 2012, 40:D303-D305.
[83] Lowe TM et al.. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 1997, 25:955-964.
[84] Altschul SF et al.. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997, 25: 3389-3402.
[85] Petersen TN et al.. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 2011, 8:785-786.
[86] Lupas A et al.. Predicting coiled coils from protein sequences. Science 1991, 252:1162-1164.
[87] Selkov E Jr et al.. MPW: the metabolic pathways database. Nucleic Acids Res 1998, 26:43-45
[88] Li L et al.. OrthoMCL. identification of ortholog groups for eukaryotic genomes. Genome Res 2003, 13: 2178-2189.
[89] Guindon S et al.. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 2010, 59:307-321.
[90] Rubber Genome Browser. http://bioinfo.ccbusm.edu.my/cgi-bin/gb2/gbrowse/Rubber/
(全文译自BMC Genomics 2013,14:75)
Draft genome sequence of the rubber treeHeveabrasiliensis
Translated by TIAN Lang
RubberResearchInstitute,ChineseAcademyofTropicalAgriculturalSciences,Danzhou,Hainan571737
Heveabrasiliensis,a member of the Euphorbiaceae family,is the major commercial source of natural rubber (NR).NR is a latex polymer with high elasticity,flexibility,and resilience that has played a critical role in the world economy since 1876.Here,we report the draft genome sequence of H.brasiliensis.The assembly spans~1.1 Gb of the estimated 2.15 Gb haploid genome.Overall,78% of the genome was identified as repetitive DNA.Gene prediction shows 68,955 gene models, of which 12.7% are unique to Hevea.Most of the key genes associated with rubber biosynthesis,rubberwood formation,disease resistance,and allergenicity have been identified.The knowledge gained from this genome sequence will aid in the future development of high-yielding clones to keep up with the ever increasing need for natural rubber.
Heveabrasiliensis; Euphorbiaceae; Natural rubber; Genome
2016-04-16
田 郎(1961-),男,侗族,湖南新晃人,硕士,副研究员,现从事植物组织培养及分子生物学研究工作。
S
A
1001-2117(2017)02-0101-15
猜你喜欢
热带生物学报(2022年1期)2022-03-09
农业科技与信息(2021年9期)2021-12-07
军事文摘(2021年18期)2021-12-02
今日农业(2021年11期)2021-08-13
小学科学(学生版)(2021年6期)2021-07-21
中西医结合肝病杂志(2020年2期)2020-10-27
中成药(2018年7期)2018-08-04
小学科学(学生版)(2018年6期)2018-06-26
贵州科学(2016年5期)2016-11-29
世界热带农业信息(2016年6期)2016-07-02
