
宏基因组方法比较分析深海和珠江口沉积物中抗生素耐药基因的特征*
2017-06-10 08:41:47林岚林琳陈恩中陈保卫王晓玮陈清
中山大学学报(自然科学版)(中英文) 2017年2期
林岚 ,林琳,陈恩中,陈保卫,王晓玮,陈清
(1. 南方医科大学公共卫生学院,广东 广州 510515; 2. 南方医科大学珠江医院,广东 广州 510282; 3. 中山大学孙逸仙纪念医院,广东 广州 510020; 4. 中山大学海洋学院,广东 广州 510275)
宏基因组方法比较分析深海和珠江口沉积物中抗生素耐药基因的特征*
林岚1, 2,林琳3,陈恩中2,陈保卫4,王晓玮4,陈清1
(1. 南方医科大学公共卫生学院,广东 广州 510515; 2. 南方医科大学珠江医院,广东 广州 510282; 3. 中山大学孙逸仙纪念医院,广东 广州 510020; 4. 中山大学海洋学院,广东 广州 510275)
细菌对抗生素的耐药性是全球关注的环境健康问题之一。该研究旨在使用宏基因组方法比较分析西太平洋深海和珠江口沉积物中抗生素耐药基因,认识抗生素使用与环境细菌耐药性间的关系。研究发现,深海沉积物多重耐药基因的含量高达77.8%;珠江口沉积物多重耐药基因只有27.2%,常用抗生素的耐药基因(磺胺类、大环内酯类、氨基糖苷类等)含量明显提高(约70%)。沉积物中共发现45种耐药基因亚型,其中7种基因亚型(acrB、amrB、bacA、ceoB、macB、mexB和smeE)能在所有沉积物中发现。深海沉积物中质粒仅携带0.3%的耐药基因,而珠江口沉积物则超过40%。研究表面,由于珠江口周边区域常用抗生素的广泛使用,其沉积物中细菌抗生素耐药性明显提高、耐药机制趋于多样化。
抗生素耐药基因;宏基因组分析;沉积物;西太平洋;珠江口
抗生素耐药基因(antibiotic resistance genes, ARGs)作为一种新兴的环境“污染物”已受到广泛的重视[1]。从人类活动干扰环境采集的水、土壤和沉积物等样品中,经常发现高丰度的多样的抗生素耐药基因[2-4]。抗生素耐药基因可以从环境细菌传播到人类病原菌,显著降低了抗生素药物的疗效,对人类健康造成严重的影响[5]。在临床治疗中,一些抗生素由于普遍的耐药性而放弃使用,给临床治疗带来很大的问题[6]。自然环境中存在大量微生物,没有人类干扰的自然环境中也可以检出抗生素耐药基因[7-9]。环境微生物可分泌抗生素以抑制其他微生物的生长,作为微生物之间的一种竞争模式,抗生素的微生物合成已经过成千上万年的进化[10]。此外,一些细菌可以将抗生素转化营养物质,支撑它们的生长[11]。无论分泌抗生素的微生物还是利用抗生素的微生物都需要携带抗生素耐药基因,保护它们免受抗生素的危害[12-14]。由于人类在医学、农业、养殖业中广泛应用抗生素,自然环境如水体和土壤中抗生素污染严重,例如,在珠江口环境中检测到较高浓度的抗生素[15-16]。然而,人类活动区域环境与没有人类干扰的自然环境中的抗生素以及耐药基因的分布差异尚不明确。
本研究将使用基于高通量DNA测序技术的宏基因组分析方法分别研究深海和珠江口沉积物中的细菌群落结构、抗生素耐药基因的种类与水平及其位置,比较分析自然环境与人类活动环境中抗生素耐药基因的特点,以探讨人类活动对周边水环境的影响。
1 材料和方法
1.1 样品采集
2014年12月于西太平洋深海(东经134°50′5.22″,北纬10°00′12.18″),水深4 042 m,采集了一个沉积物样品,样本编号WP1。2011年8月分别在珠江口的虎门(东经113°40′00″,北纬22°43′59″)和深圳湾(东经113°56′57″,北纬22°28′12″)采集了两个沉积物样品,样本编号分别为PRE1和PRE2。珠江口PRE1采样点位于狮子洋河口区域,水深20.0 m,珠江广州段及北江由此流入珠江口。珠江口PRE2采样点位于深圳湾,水深5.6 m。深圳湾位于香港和深圳市之间的一个半封闭海湾,笼箱式水产养殖活动密集。使用抓斗采集沉积物样品,采集后立即放于无菌的聚乙烯塑料袋中,放到4~6 ℃冰箱保存,直至DNA提取。
1.2 沉积物中基因组提取和高通量测序
使用FastDNA®SPIN Kit (MP Biomedicals, Santa Ana, CA)试剂盒根据产品操作手册提取沉积物中的DNA。由于海洋沉积物中DNA含量较低,需对沉积物中的DNA进行多次提取并混合,以消除样品的不均一性和单次DNA提取导致的误差。使用Thermo Scientific NanoDrop 1000 分光光度计测定DNA提取物的浓度与纯度,DNA样品都被送到安诺基因(北京,中国)进行测序。大约5 μg DNA样品被剪切成200 bp长度的片段,并使用T4DNA polymerase、Klenow Fragment和T4Polynucleotide Kinase进行末端修复。随后接头(Adapters)被连接到DNA片段,并使用凝胶电泳纯化需要的DNA片段。使用PCR扩增DNA目标片段,同时将index tags连接到接头上构建DNA文库,最后使用Illumina HiSeq 2500对构建的文库进行测序。每个沉积物样品的测序输出量约为3.0 Gbp。
1.3 生物信息学分析
首先对原始测序数据(序列长度125 bp)进行质量控制,去除含有测序错误和测序质量分数低于20的序列。通过使用BLASTN程序(Linux Release 2.2.29)与SILVA Small Subunit (SSU)数据库(version 10.4)进行比对,从测序数据中识别并抽取16S rRNA基因[17],比对门槛值E-value为10-20。随后将16S rRNA基因的比对结果输入到MEGAN软件(version 4.70.4),使用lowest common ancestor (LCA) algorithm算法,绝对截止值为BLAST bitscore 等于50,相对截止值为50个比对结果的10%,分析沉积物中的细菌群落结构[18]。将测序数据与antibiotic resistance genes database (ARDB)数据库比对[19],如果比对长度大于25个氨基酸、相似度大于90%,该序列被认为是耐药基因[20]。此外,根据上传到NCBI数据库中的参考序列,建立一个质粒基因数据库[20]。如果比对长度大于90个碱基、相似度大于95%,该序列被识别为质粒基因。
2 结果与讨论
2.1 西太平洋和珠江口沉积物中细菌群落结构
宏基因测序数据首先与SSU微生物分类数据库进行比对,获取16S rRNA基因,然后用MEGAN软件分析16S rRNA基因的比对结果,以获取沉积物中细菌群落结构信息。结果如图1所示,沉积物中的细菌分布在16个菌门,不管在西太平洋还是在珠江口的沉积中,多数细菌属于变形菌门(Proteobacteria)。但是,西太平洋沉积物中变形菌门的细菌相对百分比高达95%,远远高于其在珠江口沉积物中的比例。显然,西太平洋和珠江口沉积物中细菌群落结构存在明显差异。在珠江口区域,咸淡水交汇,因此淡水和海水微生物在口门区域交汇,而在西太平洋深海主要是能在极端环境下(如低温、营养物质匮乏)生存的微生物。
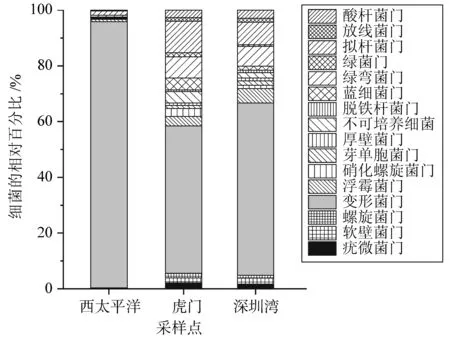
图1 西太平洋和珠江口沉积物中细菌群落结构的比较Fig.1 Comparison of bacterial community structures between the WP and PRE sediments
2.2 西太平洋和珠江口沉积物中抗生素耐药基因的丰度和耐药性特征
沉积物中识别的抗生素耐药基因根据抗生素的种类进行分类,即耐药性类型。此外,样品中抗生素耐药基因的丰度使用16S rRNA基因进行归一化处理,结果如图2所示。抗生素耐药基因在珠江口PRE1处采集的沉积物中丰度最高,依次为西太平洋及珠江口PRE2采样点。沉积物中各抗性类型的抗生素耐药基因的相对组成如图3所示。在西太平洋沉积物中,多重耐药基因和氯霉素耐药基因相对含量最高,特别是多重耐药基因(multidrugs resistance genes)高达77.8%。与西太平洋沉积物相比,珠江口沉积物中多重耐药基因的相对含量较低;相反,与常用抗生素(如,磺胺类、大环内酯类、氨基糖苷类、β-内酰胺类等)相关的耐药基因的相对含量则明显提高。
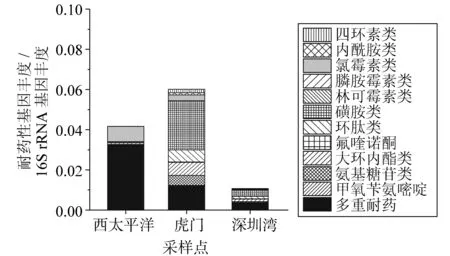
图2 西太平洋和珠江口沉积物中抗生素耐药性基因的比较Fig.2 Abundance and resistance types of ARGs in the sediments of the in the WP and PRE sediments

图3 西太平洋和珠江口沉积物中抗生素耐药性基因的Fig.3 Relative percentages of ARG resistance types in the WP and PRE sediments
在无污染的自然环境中发现抗生素耐药基因,表明了耐药基因的自然起源[7-9]。然而由于耐药基因的极大的多样性,很难获取环境介质中耐药基因的综合信息和特征。近来高通量测序技术和宏基因组分析技术的迅速发展[21],帮助准确识别环境介质中的耐药基因的综合特征[22]。在西太平洋深海沉积物中,没有污染物胁迫的条件下细菌形成并维持了独特的抗生素耐药基因特征,以多重耐药基因为主,少有针对常用抗生素的特异性耐药基因。但是,由于抗生素在珠江口周边地区的大量使用,抗生素、耐药菌及其携带的耐药基因从典型污染源(如污水处理厂、家禽养殖场、水产养殖区等)排放到河口环境中,因而珠江口沉积物中耐药基因特征以常用抗生素的特异性基因为主。例如,先前的研究表明,四环素和磺胺耐药基因的水平,从珠江内河道、珠江口到近海区域呈明显降低趋势,耐药基因的水平与抗生素浓度显著相关[4, 23]。这些研究恰恰与我们的结果一致,在珠江口沉积物中发现大量的四环素和磺胺耐药基因而在深海沉积物中则没有。
2.3 西太平洋和珠江口沉积物中抗生素耐药基因的亚型
在西太平洋和珠江口沉积物中总共发现45种抗生素耐药基因亚型。其中7种抗生素耐药基因亚型能在所有样品中被发现,包括acrB、amrB和mexB是多重耐药基因,bacA是环肽耐药基因,ceoB是氯霉素耐药基因,macB是大环内酯耐药基因,smeE是喹诺酮耐药基因,这些耐药基因亚型在沉积物中检测到的同类耐药基因中大多数都是丰度最高的。例如,在所有沉积物中ceoB占总氯霉素耐药基因的至少70%。区别于西太平洋,氨基糖苷主要耐药基因亚型(aad9和ant2IA)、β-内酰胺主要耐药基因亚型(bl2d_oxa2和bl3_imp)磺胺主要耐药基因亚型(sul1)只在珠江口沉积物中发现,说明由于这些抗生素的大量使用,导致污染环境中的细菌进化并富集针对这些常用抗生素的独特耐药基因亚型(图4)。

图4 西太平洋和珠江口沉积物中抗生素耐药性基因亚型Fig.4 Venn diagram of ARG subtypes in the WP and PRE sediments
2.4 西太平洋和珠江口沉积物中质粒基因丰度和抗生素耐药基因的水平传播
通过与质粒数据库比对,分析了西太平洋和珠江口沉积物中质粒基因的丰度,结果如图5所示。所有采集的样品中,在WP1采集的深海沉积物的质粒基因丰度最高,其丰度要比珠江口沉积物至少高5倍。同时,从测序数据中提取抗生素耐药基因序列,并将其与质粒数据库进行比对,以分析质粒携带的耐药基因的相对含量。图5显示,质粒携带的抗生素耐药基因的趋势与质粒基因丰度完全相反,珠江口沉积物中超过40%的耐药基因在质粒上被发现,而西太平洋沉积物中仅仅0.3%的耐药基因在质粒上。结果表明,珠江口沉积物中抗生素耐药基因的水平传播潜势要远远高于西太平洋深海沉积物中的耐药基因。

图5 西太平洋和珠江口沉积物中质粒基因丰度和质粒携带的耐药基因的相对含量Fig.5 Abundance of plasmid genes and relative percentages of plasmid-related ARGs in the WP and PRE sediments
自然环境中的大多数微生物对抗生素具有较低的耐药性,其携带质粒基因组基含低水平的抗生素耐药基因[24-25],这与我们在西太平洋深海沉积物的宏基因分析结果完全一致。此外,转化实验也表明自然环境中的耐药基因不能在细菌之间传递[26]。然而,随着人类干扰环境中抗生素胁迫的提高,导致耐药基因通过质粒介导的基因水平传递在细菌宿主之间传播[27]。因此,质粒作为抗生素耐药基因水平传播的载体,在环境中发现耐药基因丰度和质粒基因丰度显著相关[3]。宏基因组分析发现珠江口沉积物中高比例的耐药基因出现在质粒上,这可能是耐药基因在细菌间水平传播的结果,也必将有利于抗生素耐药性在细菌间的继续传播。
3 结 论
基于高通量DNA测序技术的宏基因组分析方法,能够综合评价西太平洋和珠江口沉积物中的抗生素耐药基因的整体特征。在西太平洋和珠江口沉积物之间抗生素耐药基因的丰度和种类都存在显著差异,说明在抗生素广泛、大量使用的情况下,人类严重干扰环境中细菌对抗生素耐药性的进化已与自然环境产生分化,其耐药基因主要以表达对常用抗生素(四环素、磺胺、氨基糖苷、β-内酰胺等)的耐药性为主。与深海沉积物相比,珠江口沉积物含有更高比例的质粒携带的抗生素耐药基因,表面质粒介导的基因水平传递是细菌获取抗生素耐药性的主要方式。
[1]PRUDENA,PEIR,STORTEBOOMH,etal.Antibioticresistancegenesasemergingcontaminants:studiesinnorthernColorado[J].EnvironmentalScience&Technology, 2006, 40: 7445-7450.
[2]KNAPPCW,DOLFINGJ,EHLERTPA,etal.Evidenceofincreasingantibioticresistancegeneabundancesinarchivedsoilssince1940[J].EnvironmentalScience&Technology, 2010, 44: 580-587.
[3]CHENBW,YANGY,LIANGXM,etal.Metagenomicprofilesofantibioticresistancegenes(ARGs)betweenhumanimpactedestuaryanddeepoceansediments[J].EnvironmentalScience&Technology, 2013, 47: 12753-12760.
[4]CHENBW,LIANGXM,HUANGXP,etal.DifferentiatinganthropogenicimpactsonARGsinthePearlRiverEstuarybyusingsuitablegeneindicators[J].WaterResearch, 2013, 47: 2811-2820.
[5]PEHRSSONEC,TSUKAYAMAP,PATELS,etal.Interconnectedmicrobiomesandresistomesinlow-incomehumanhabitats[J].Nature, 2016, 533: 212-216.
[6]WENZELRP,EDMONDMB.Managingantibioticresistance[J].NewEnglandJournalofMedicine, 2000, 343: 1961-1963.
[7]D’COSTAVM,KINGCE,KALANL,etal.Antibioticresistanceisancient[J].Nature, 2011, 477: 457-461.
[8]BHULLARK,WAGLECHNERN,PAWLOWSKIA,etal.Antibioticresistanceisprevalentinanisolatedcavemicrobiome[J].PLoSOne, 2012, 7:e34953.
[9]BROWNMG,BALKWILLDL.Antibioticresistanceinbacteriaisolatedfromthedeepterrestrialsubsurface[J].MicrobialEcology, 2009, 57: 484-493.
[10]BALTZRH.Renaissanceinantibacterialdiscoveryfromactinomycetes[J].CurrentOpinioninPharmacology, 2008, 8: 557-563.
[11]DANTASG,SOMMERMOA,OLUWASEGUNRD,etal.Bacteriasubsistingonantibiotics[J].Science, 2008, 320: 100-103.
[12]CORDEROOX,WILDSCHUTTEH,KIRKUPB,etal.Ecologicalpopulationsofbacteriaactassociallycohesiveunitsofantibioticproductionandresistance[J].Science, 2012, 337: 1228-1231.
[13]WRIGHTGD.Theantibioticresistome:thenexusofchemicalandgeneticdiversity[J].NatureReviewsMicrobiology, 2007, 5: 175-186.
[14]WRIGHTGD.Theantibioticresistome[J].ExpertOpiniononDrugDiscovery, 2010, 5: 779-788.
[15]XUWH,ZHANGG,LIXD,etal.OccurrenceandeliminationofantibioticsatfoursewagetreatmentplantsinthePearlRiverDelta(PRD),SouthChina[J].WaterResearch, 2007, 41: 4526-4534.
[16]LIANGX,CHENB,NIEX,etal.ThedistributionandpartitioningofcommonantibioticsinwaterandsedimentofthePearlRiverEstuary,SouthChina[J].Chemosphere, 2013, 92: 1410-1416.
[17]ALTSCHULSF,MADDENTL,SCHAFFERAA,etal.GappedBLASTandPSI-BLAST:anewgenerationofproteindatabasesearchprograms[J].NucleicAcidsResearch, 1997, 25: 3389-3402.
[18]HUSONDH,AUCHAF,QIJ,etal.MEGANanalysisofmetagenomicdata[J].GenomeResearch, 2007, 17: 377-386.
[19]LIUB,POPM.ARDB-antibioticresistancegenesdatabase[J].NucleicAcidsResearch, 2009, 37:D443-D447.
[20]YANGY,LIB,JUF,etal.Exploringvariationofantibioticresistancegenesinactivatedsludgeoverafour-yearperiodthroughametagenomicapproach[J].EnvironmentalScience&Technology, 2013, 47: 10197-10205.
[21]MUNCKC,ALBERTSENM,TELKEA,etal.Limiteddisseminationofthewastewatertreatmentplantcoreresistome[J].NatureCommunications, 2015, 6: 8452.
[22]LIB,YANGY,MAL,etal.Metagenomicandnetworkanalysisrevealwidedistributionandco-occurrenceofenvironmentalantibioticresistancegenes[J].ISMEJ, 2015, 9: 2490-2502.
[23]CHENBW,LIANGXM,NIEXP,etal.TheroleofclassIintegronsinthedisseminationofsulfonamideresistancegenesinthePearlRiverandPearlRiverEstuary,SouthChina[J].JournalofHazardousMaterials, 2015, 282: 61-67.
[24]DAVISCE,ANANDANJ.Theevolutionofrfactor.Astudyofa“preantibiotic”communityinBorneo[J].NewEnglandJournalofMedicine, 1970, 282: 117-122.
[25]HUGHESVM,DATTAN.Conjugativeplasmidsinbacteriaofthe“pre-antibiotic”era[J].Nature, 1983, 302: 725-726.
[26]FREDRICKSONJK,HICKSRJ,LISW,etal.Plasmidincidenceinbacteriafromdeepsubsurfacesediments[J].AppliedandEnvironmentalMicrobiology, 1988, 54: 2916-2923.
[27]BELLANGERX,GUILLOTEAUH,BONOTS,etal.Demonstratingplasmid-basedhorizontalgenetransferincomplexenvironmentalmatrices:apracticalapproachforacriticalreview[J].ScienceoftheTotalEnvironment, 2014, 493: 872-882.
Metagemomic analysis on characteristics of antibiotic resistance genes in the deep ocean and Pearl River Estuary sediments
LINLan1, 2,LINLin3,CHENEnzhong2,CHENBaowei4,WANGXiaowei4,CHENQing1
(1. School of Public Health, Southern Medical University, Guangzhou 510515, China; 2. Zhujiang Hospital of Southern Medical University, Guangzhou 510282, China; 3. Department of Respiratory Disease, Sun Yat-sen Memorial Hospital, Guangzhou 510020, China; 4. School of Marine Science, Sun Yat-sen University, Guangzhou 510275, China)
Antibiotic resistance is one of the most important environmental issues in the world. The relationship between antibiotic use and antibiotic resistance of environmental bacteria were investigated using metagenomic analysis of antibiotic resistance genes (ARGs) in the sediments collected from the Western Pacific (WP) and the Pearl River Estuary (PRE) in this work. The result indicated that multidrug resistance genes (MRGs) were accounted as 77.8% of the total ARGs in the WP sediments, and the PRE sediments as 27.2%. ARGs in the PRE sediments were characterized by increasing abundance of ARGs specific to common antibiotics (e.g., sulfonamides, macrolides, aminoglycoside, etc.) and decreasing relative percentage of MRGs. A total of 45 ARG subtypes were identified in the sediments, and thereof 7 subtypes, includingacrB,amrB,bacA,ceoB,macB,mexB andsmeE, concurrently existed in all sediments. Approximately 0.3% of ARGs in the WP sediments were potentially carried by the plasmids. Different from the WP, relative percentage of plasmid-carrying ARGs in the PRE sediments was higher than 40%. It was suggested that the abundance and diversity of ARGs in the PRE environment were substantially increased probably due to wide use of antibiotics in the nearby regions.
antibiotic resistance genes; metagenomic analysis; sediments; Western Pacific; Pearl River Estuary
2016-11-06 基金项目:广东省自然科学基金(2014A030313195)
林岚(1982年生),女;研究方向:感染性疾病流行病学;E-mail:105824412@qq.com
陈清(1959年生),女;研究方向:感染性疫病流行病学、评价流行病学;E-mail:qch.2009@163.com
10.13471/j.cnki.acta.snus.2017.02.018
R11
A
0529-6579(2017)02-0112-05
猜你喜欢
海洋通报(2022年2期)2022-06-30 06:07:04
水文地质工程地质(2022年2期)2022-04-13 09:02:02
海洋石油(2021年3期)2021-11-05 07:43:12
河北环境工程学院学报(2021年1期)2021-03-19 08:43:00
新城乡(2018年11期)2018-11-22 03:11:02
西藏科技(2016年5期)2016-09-26 12:16:42
海洋渔业(2016年6期)2016-04-16 03:00:37
环境科技(2015年4期)2015-11-08 11:10:44
中国海洋大学学报(自然科学版)(2014年8期)2014-02-28 12:21:28
