
1,8—萘二甲酰亚胺自由基清除活性相关量子化学参数的计算研究
2017-05-30 06:19苏聪邹碧群
科技风 2017年11期
苏聪 邹碧群
摘要:结合Hyperchem软件,以从头算法、半经验算法和分子力场算法等三种算法对1,8萘二甲酰亚胺的结构进行优化计算,将计算结果与单晶实测数据进行比较,根据计算结果与晶体实测数据的吻合度推断出半经验算法是最适合计算1,8萘二甲酰亚胺结构的算法,最后进一步基于半经验算法对1,8萘二甲酰亚胺自由基清除活性相关的量子化学参数进行计算。结果表明,净电荷的分布、HOMO能量和LUMO能量及其电子云的排布情况是影响1,8萘二甲酰亚胺自由基清除能力的主要指标。
关键词:1,8萘二甲酰亚胺;自由基清除活性;AM1半经验方法;量子化学参数
中图分类号:G641文献标识码:A
计算化学是根据量子化学、统计热力学及经典力学及大量的数值运算方式研究分子、团簇的性质及化学反应的一门科学[12],其有效地将计算机技术与量子化学集成一体,旨在通过理论计算相关量子化学参数,总结或预见化学物质與其性能之间的规律[3]。随着近二十年来计算机技术的高速发展,计算化学获得了迅猛发展,而伴随计算化学家Walter Kohn与John Pople斩获1998年的诺贝尔化学奖后,计算化学愈发受到人们的高度关注,通过计算化学来解释和解决化学、物理、分子生物和材料等方面的问题逐渐成为一种行之有效的手段[4]。
自由基清除剂是指能清除自由基或能阻断自由基参与氧化反应的物质,其具有延缓衰老、防治癌症等重要功效,对维持机体的正常生命活动和保持机体健康起着重要的作用[5]。因此,自由基清除剂的计算设计研究受到学者的高度关注。在我们的前期研究中,我们首次报道1,8萘二甲酰亚胺具有良好的自由基清除活性[6]。作为研究的延续,本文主要采用Hyperchem软件计算1,8萘二甲酰亚胺自由基清除活性的相关量子化学参数,从理论和结构上阐述其自由基清除活性的根源,为进一步理性设计1,8萘二甲酰亚胺自由基清除剂提供理论参考。
1 材料与方法
1.1 仪器与设备
Hyperchem 8.0计算软件、PC计算机、Bruker SMART APEX2 CCD单晶衍射仪。
1.2 量子化学与分子力学计算方法
以 Chemoffice 软件中的 Chemdraw 程序画出1,8萘二甲酰亚胺的基本结构,导入Hyperchem 8.0软件后,分别采用从头算法、半经验算法和分子力场算法等三种算法对1,8萘二甲酰亚胺的结构进行优化,将计算结果(表1)与1,8萘二甲酰亚胺的单晶数据(表1)进行比较,通过比较计算结果和晶体实测数据的吻合度后,推断出半经验算法是比较适合1,8萘二甲酰亚胺结构的算法,并进一步用半经验方法AM1模式计算1,8萘二甲酰亚胺中各原子的键长、键角与净电荷分布、最高已占轨道(HOMO)和最低未占轨道(LUMO)的能量和电子云排布等量子化学参数。
1.3 1,8萘二甲酰亚胺晶体的制备与衍射数据收集
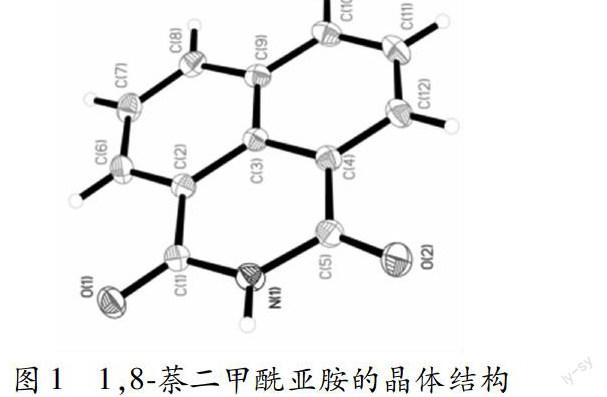
将0.198g 1, 8萘酐與10mL氨水混合于50mL的二氯甲烷乙醇溶液中(体积比为1:2),80oC下回流4h,冷却,得到0.19g 1,8萘二甲酰亚胺晶体,产率92.95%。晶体结构(图1)和数据(表1)采用Bruker SMART APEX2 CCD单晶衍射仪收集。由图1可见,1,8萘二甲酰亚胺的结构得到明确的表征。
2 结果与讨论
2.1 三种算法对1,8萘二甲酰亚胺结构的适合性比较
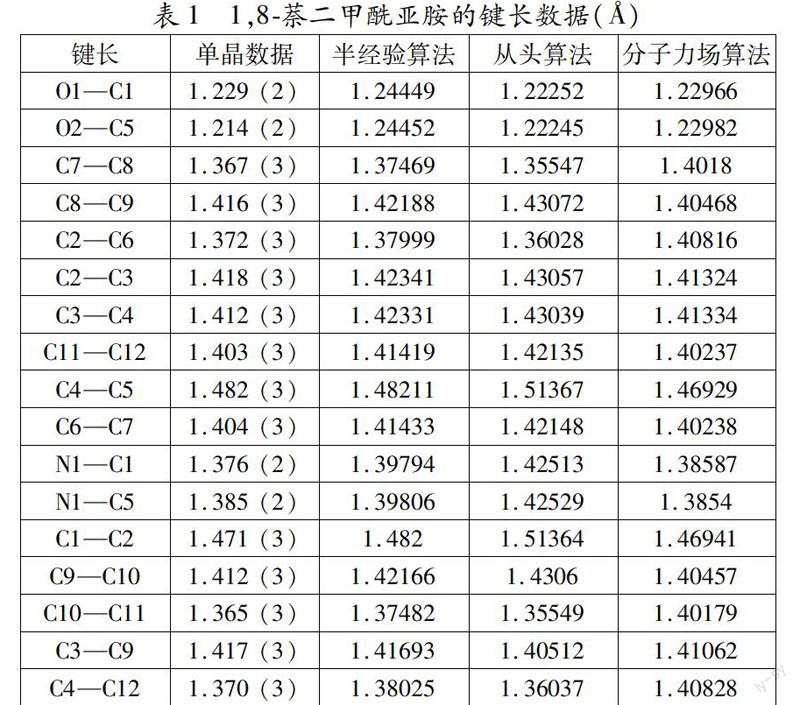
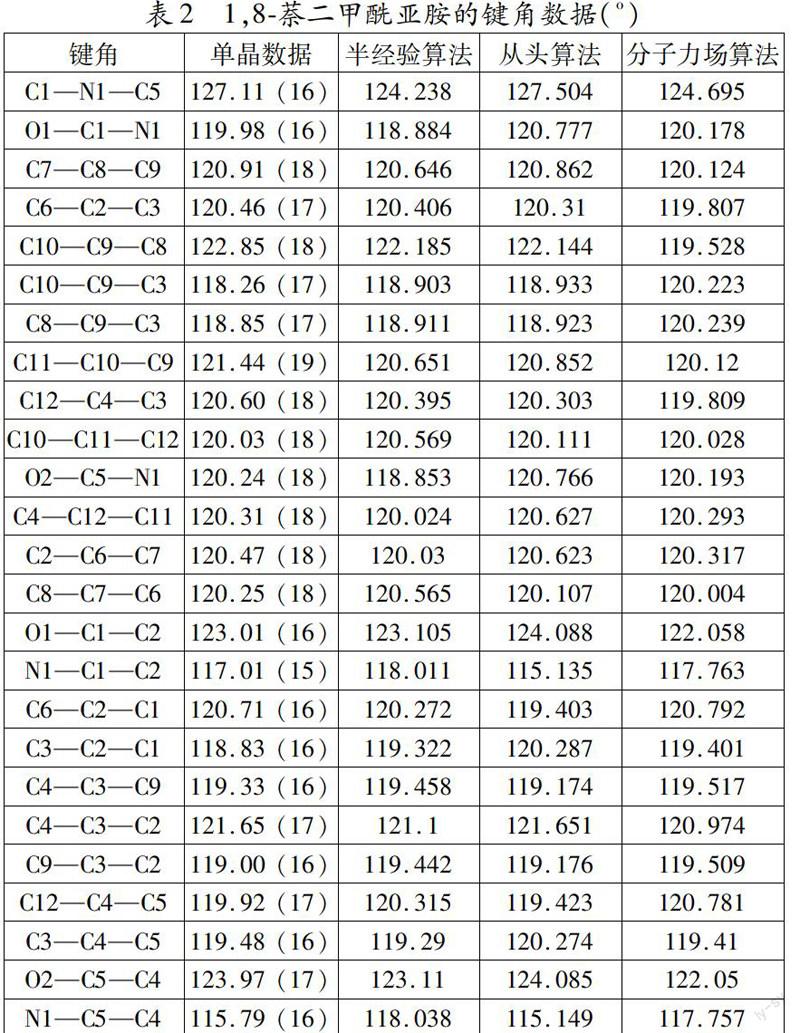
本文采用从头算法、半经验算法和分子力场算法分别优化1,8萘二甲酰亚胺结构,然后再把每个算法得到的基本结构参数与其晶体结构的实测参数进行比较,找出最接近晶体实测参数的算法,以此判断哪种算法最适合1,8萘二甲酰亚胺的结构优化计算。具体的计算结果和晶体数据分别列于表1和表2中。
由表1可见,采用半经验算法计算得到的1,8萘二甲酰亚胺的键长与通过晶体数据收集的键长真实值最接近,只有N1—C1的键长值具有较大的误差,误差值为0.03052;采用分子力场算法计算得到的1,8萘二甲酰亚胺的键长与通过晶体数据收集的键长真实值的误差较小,其中C7—C8、C2—C6、C10—C11和C4—C12的键长的误差值分别为0.0348、0.03616、0.03679和0.03828;而从头算法计算得到的1,8萘二甲酰亚胺的键长与通过晶体数据收集的键长真实值的误差最大,其中C4—C5、N1—C1、N1—C5和C1—C2的键长的误差值分别为0.03167、0.04913、0.04029和0.04264。由此可以判断,三种算法的适合性顺序为:半经验算法 > 分子力场法 > 从头算法。
由表2可见,以从头算法计算得到的1,8萘二甲酰亚胺的键角与通过晶体数据收集的键角真实值最接近,只有O1—C1—C2、N1—C1—C2、C6—C2—C1和C3—C2—C1的键角值具有较大的误差,误差值分别为1.078 o、1.875 o、1.307 o和1457 o;采用从半经验算法计算得到的1,8萘二甲酰亚胺的键角与通过晶体数据收集的键角真实值较为接近,其中C1—N1—C5、O1—C1—N1、O2—C5—N1和N1—C5—C4的键角值具有较大的误差,误差值分别为2.872 o、1.096 o、1.387 o、1.001 o和2.248 o;而采用从半经验算法计算得到的1,8萘二甲酰亚胺的键角与通过晶体数据收集的键角真实值误差最大,C1—N1—C5、C10—C9—C8、C8—C9—C3、C11—C10—C9、O2—C5—C4和N1—C5—C4的键长值具有较大的误差,误差值分别为2.415 o、3.322 o、1.389 o、1.32 o、1.92 o和1.967 o。由此可以判断,三种算法的适合性顺序为:从头算法 > 半经验算法 > 分子力场法。
最后,综合以上两种结果评估可知,三种算法对1, 8萘二甲酰亚胺结构的适合性顺序为:半经验算法 > 从头算法 > 分子力场法,该结果与钱旭红课题组的报道一致[7]。
萘二甲酰亚胺各原子的净电荷分布情况
自由基具有良好的反应活性,容易与带电负性的原子或分子发生结合,从而发生自由基清除作用[7]。因此,通过计算自由基清除剂的原子的净电荷分布情况,可为判断其自由基清除活性提供依据。
本文采用半经验算法对1,8萘二甲酰亚胺的原子的净电荷分布情况进行了计算,计算结果列于表3。由表3可见,只有N1、C1和C5共3个原子带有电正性,其余的12原子均展示为电负性,说明1,8萘二甲酰亚胺与自由基反应的原子点较多,亲核性较强,容易与自由基发生反应,从而可能导致其具有较好的自由基清除活性,这与我们之前的报道相吻合[6]。
2.3 HOMO和LUMO的计算
研究表明,自由基清除活性與HOMO和LUMO的能量有密切关系,HOMO的能量越高,分子的供电子(推电子)能力越好,自由基清除活性则越高[89],LUMO的能量越低,分子的接受电子(拉电子)能力越强。为了从结构上进一步验证1,8萘二甲酰亚胺自由基清除活性的根源,本文还进一步计算了其HOMO和LUMO的能量和电子排布情况。
由表4的计算结果可见,1,8萘二甲酰亚胺HOMO能量值较高,表明其给电子的能力较强,有利于形成推拉电子效应,从而可能导致其具有较好的自由基清除活性,这与之前的报道相一致[6,7]。
图2是1,8萘二甲酰亚胺的HOMO和LUMO电子排布情况,图中用分子骨架上的红色网格线来表示推电子,拉电子用绿色显示。由图可见,1,8萘二甲酰亚胺可以形成良好的推拉电子效应,从而有利于电子的流动,这可能导致其具有较好自由基清除活性的原因之一。
3 结论
综合以上计算结果可知:半经验算法较适合1,8萘二甲酰亚胺结构的计算;1,8萘二甲酰亚胺的自由基清除活性可能主要与净电荷分布、HOMO和LUMO的能量和电子排布等参数有关。
参考文献:
[1]里沃斯.计算化学:分子和量子力学理论及应用导论[M].科学出版社,2012.
[2]黄晓峰,曹同成,陈波.计算化学——探索化学世界的罗盘[J].化学教学,2014,3:1113.
[3]苑世领,张恒,张冬菊.分子模拟:理论与实验[M].化学工业出版社,2016.
[4]蘇培峰,谭凯,吴安安,等.理论与计算化学研究进展[J].厦门大学学报(自然科学版),2011,50(3):311318.
[5]Ye Zhang, BiqunZou, Zhenfeng Chen, et al, Synthesis and antioxidant activities of novel 4Schiff base7benzyloxycoumarin derivatives[J]. Bioorganic & Medicinal Chemistry Letters, 2011,21:68116815.
[6]Ye Zhang, Shaobo Feng, Qiang Wu, etal, Microwaveassisted synthesis and evaluation of naphthalimides derivatives as free radical scavengers[J].Medicinal Chemistry Research,2011,20:752759.
[7]Zhigang Li, Qing Yang, Xuhong Qian, Novel thiazonaphthalimides as efficient antitumor and DNA photocleaving agents: Effects of intercalation, side chains, and substituent groups[J].Bioorganic & Medicinal Chemistry,2005,13:48644870.
[8]刘科梅,聂挺,潘栋梁,等.量子化学计算研究4种黄酮类天然抗氧化物清除 自由基活性的构效关系[J].南昌大学学报(理科版),2016,40(3):250256.
[9]苏彦雷,张骥,蒋建勤,等.量子化学计算法分析蒙古黄芪中部分黄酮类化合物 抗氧化作用机制[J].中国药科大学学报,2011,42(1): 3943.
基金项目:广西自然科学基金项目(2016GXNSFAA380300、2014GXNSFBA118050)
作者简介:苏聪(1982),女,汉族,广西贵港人,硕士,桂林师范高等专科学校教师。
猜你喜欢
高中数理化(2022年16期)2022-09-14
民用飞机设计与研究(2020年1期)2020-05-21
青岛大学学报(工程技术版)(2019年2期)2019-09-10
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22
中国塑料(2017年2期)2017-05-17
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16
中学化学(2015年8期)2015-12-29
中国塑料(2015年6期)2015-11-13
郑州大学学报(理学版)(2014年4期)2014-03-01
海南师范大学学报(自然科学版)(2013年2期)2013-10-12
