
儿童Alstrom综合征1例报告及文献复习
2017-05-25 00:37蔡清霞常国营丁宇李娟程青李辛王剑王秀敏沈亦平
临床儿科杂志 2017年4期
蔡清霞常国营丁 宇李 娟程 青李 辛王 剑王秀敏沈亦平
1.福建省龙岩市中医院(福建龙岩 364000);2.上海交通大学医学院附属上海儿童医学中心(上海 200127)
儿童Alstrom综合征1例报告及文献复习
蔡清霞1常国营2*丁 宇2李 娟2程 青2李 辛2王 剑2王秀敏2沈亦平2
1.福建省龙岩市中医院(福建龙岩 364000);2.上海交通大学医学院附属上海儿童医学中心(上海 200127)
目的分析罕见的Alstrom综合征的临床特征,及诊断和治疗。方法回顾1例Alstrom综合征患儿的临床资料及二代测序检测ALMS1基因分析结果,并复习相关文献。结果12岁10个月的女性患儿,自出生1个月余患扩张性心肌病,之后逐渐出现、肥胖、视神经疾患、感音神经性听力下降、血糖偏高、月经不规则。实验室检查显示,存在高雄激素水平、高血糖、高血脂、脂肪肝表现。高通量测序分析证实存在ALMS1基因突变,c.5418delC,p.Y1807Tfs*23的杂合移码变异,c.10549C>T,p.Q3517*的杂合无义变异;其中,c.5418delC为首次报道的新变异。结论Alstrom综合征为常染色体隐性遗传性疾病,主要表现为多脏器功能减退、代谢综合征等,可通过基因检测确诊。
Alstrom综合征; ALMS1基因; 基因突变; 遗传咨询
Alstrom 综合征,由ALSM1基因突变导致,属常染色体隐性遗传性疾病。临床主要有视力减退、神经性耳聋、肥胖、糖尿病、尿崩症、肾功能不全、性腺功能低下、高尿酸血症及高三酰甘油血症等,1959年 由Alstrom 等[1]首次报道。迄今已有500余例报道,而国内仅为个例报道[2-8]。本研究分析上海儿童医学中心收治的1例经基因分析确诊为Alstrom 综合征患儿的临床资料,同时结合国内外文献,探讨该病的主要临床特征及诊断和治疗要点。
1 临床资料
患儿,女,年龄 12岁10个月。出生后1个月余患扩张型心肌病,长期口服地高辛、卡维地洛、卡托普利,病初予泼尼松治疗半年,每半年复查彩色多普勒超声心动图,无异常。2岁余始患“视神经疾患”。3年前诊断“感音神经性听力下降”。近3年体质量增长较前明显。2年前确诊为“2型糖尿病”,予二甲双胍口服,血糖控制可。12岁时月经初潮,最初4个月内共有月经史3次,周期及行经时间不规则,后至今未再来月经。患儿系G1P1,母孕史、出生史无异常。否认父母近亲婚配。父亲有高脂血症,爷爷有糖尿病史,奶奶有原发性高血压史,母亲否认高血压、糖尿病、高脂血症及痛风等病史。同卵双胎妹妹,49天时因“扩张性心肌病合并呼吸道感染”夭折。
入院体格检查:肥胖体型,身高156 cm,体质量77 kg,体质指数(BMI)31.6 kg/m2;体温36.4℃,脉搏90次/min,呼吸24次/min,收缩压(SBP)/舒张压(DBP):119/64 mmHg,全身皮肤较黑,面部痤疮明显,眼睑无水肿,双瞳孔等圆等大,对光反射正常。颈部、腋下、腹股沟、背部、腹部、足背等可见黑棘皮,右下腹2 cm×3 cm、右腿内侧0.5 cm×1.0 cm牛奶咖啡斑各一处。双乳Tanner分期为B3期,左侧乳核2.5 cm ×2.5 cm,右侧乳核2.5 cm×2.5 cm。心率90次/min,心音稍钝,未及明显杂音。腹软,腹部皮下脂肪厚,肝脾肋下未触及,肾区无叩击痛。外阴饱满,可见阴毛,Tanner分期为PH4,可见分泌物。部分临床特征见图1。
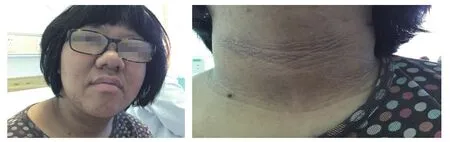
图1 患儿部分临床特征
入院实验室检查,天冬氨酸氨基转移酶 104 IU/L(10~40 IU/L),谷氨酸氨基转移酶177 IU/L(10~42 IU/ L),谷氨酰转肽酶 141 U/L(1~24 IU/L);总胆固醇5.63 mmol/L(<5.2 mmol/L),三酰甘油2.55 mmol/ L(<1.7 mmol/L),高密度脂蛋白胆固醇0.79 mmol/L (>0.91mmol/L)。肾功能、尿微量蛋白、24 h尿蛋白定量无异常。甲状腺功能、17α羟孕酮、肾素、血管紧张素、促肾上腺皮质激素、皮质醇、人绒毛膜促性腺激素、血氨、乳酸、甲胎蛋白无异常;脱氢表雄酮253 μg/dL(8.6~169.8 μg/dL);血尿串联质谱及染色体报告均未见异常;黄体生成素释放激素(LHRH )激发试验,测定0、30、60、90、120 min雌二醇(E2)、睾酮(T)、促卵泡刺激素(FSH)及黄体生成素(LH),结果提示青春发育启动,见表1。口服葡萄糖耐量试验(OGTT)示存在糖尿病,胰岛素抵抗,见表2。
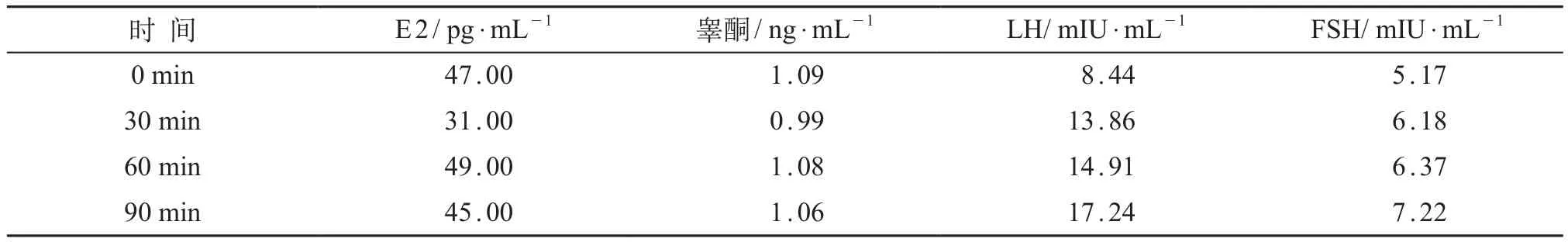
表1 患儿LHRH激发试验结果

表2 患儿OGTT试验结果
腹腔超声示脂肪肝表现,弥漫性病变待排;胰腺偏大;双肾增大。垂体磁共振成像(MRI)示垂体形态稍饱满(最厚处约7.0 mm)。彩色超声心动图示扩张型心肌病随访,左房、左室增大[LA(左房内径)=3.22 cm,LVDD(左心室舒张末期内径)=6.00 cm,12岁正常参考值上限:LA 2.63 cm,LVDD 4.66 cm],二尖瓣轻度反流,左心收缩功能稍低下。
在获得知情同意后,抽取患儿的静脉血2 mL。采用全外显子组测序方法。用德国Qiagen的QIAamp Blood DNA Mini kit试剂盒提取DNA。3 μg的基因组DNA经美国Covaris公司的M220打断仪处理后得到150~200 bp的DNA片段,采用美国Agilent公司SureSelect试剂盒建库,最后使用美国Illumina公司HiSeq 2500 System进行高通量测序。测序数据经Illumina Sequence Control Software评估合格后,使用美国SoftGenetics公司的NextGENe®软件进行数据读取,读取完毕后的数据上传到美国Ingenuity Systems公司的Ingenuity®Variant Analysis™ 软件进行生物信息学分析。
利用Sanger测序验证由上述二代测序发现的目的基因。其引物设计使用UCSC在线软件(http://genome. ucsc.edu/)。使用TaKaRa 公司的rTaq DNA聚合酶进行聚合酶链反应(PCR),PCR产物经1%的琼脂糖凝胶电泳确认后使用Qiagen 公司的QIAquick Gel Extraction Kit试剂盒纯化,纯化产物经美国Applied Biosystems公司的ABI3730XL测序仪Sanger测序,测序数据使用美国SoftGenetics公司MutationSurveyor软件进行分析。
基因检测发现该患儿ALMS1基因存在复合杂合变异:①c.5418delC,p.Y1807Tfs*23,移码变异,杂合;② c.10549C>T;p.Q3517*,无义变异,杂合。见图2。因父母拒绝留标本,且患儿为独女,未能验证是否家族遗传。
目前该患儿仍在内分泌及心内科门诊随访中,以对症治疗为主,口服药物改善心功能、减轻胰岛素抵抗,配镜改善视力,佩戴助听器改善听力等处理,可加用保肝、降脂类药物,适当增加运动、控制饮食。
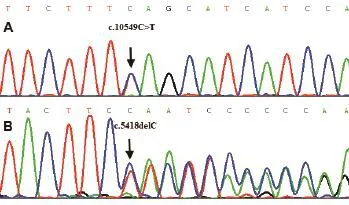
图2 患儿ALMS1基因测序图
2 讨论
Alstrom综合征人群发病率不足百万分之一[9],无性别差异,常见于近亲婚配[10]。临床表现为一系列系统纤维化和多器官受累,临床症状主要为视力减退(锥杆视网膜发育不良)、神经性耳聋、肥胖、糖尿病、尿崩症、肾功能不全、性腺功能低下、高尿酸血症及高三酰甘油血症等[11-12]。临床表现根据年龄不同表现各异。婴儿期出现视网膜变性,部分病例因扩张型心肌病而表现为充血性心力衰竭;儿童期表现为听力下降、肥胖、高胰岛素血症和2型糖尿病;青春期多为高脂血症、糖尿病及扩张型心肌病。因反复呼吸道感染,逐渐进展为肺纤维化、慢性阻塞性呼吸综合征等[12]。另有为肝纤维化、肝硬化及肾脏衰竭等表现,几乎所有器官最终都会出现纤维化病变[12]。部分变异型还可出现身材矮小、脱发、脊柱侧突等[13]。另外,该类患者可合并多种内分泌激素紊乱,包括甲状腺功能减退症,胰岛素样生长因子系统的改变,男性睾酮水平低和女性的高雄激素血症[14]。
目前Alstrom综合征通用的临床诊断标准:①生后第一年出现视网膜病变和肥胖;②七个主要标准(感音神经性聋,扩张型心肌病,2型糖尿病,肺、肝、尿异常,肾功能不全)至少符合三条;③没有并指和智力低下[9]。但新研究结果表明,按照该标准,不典型病例会漏诊[15]。本例患儿1月龄即发病,表现为扩张型心肌病、视力减退、肥胖、感音神经性听力下降等症状。辅助检查提示2型糖尿病、高雄激素血症、高脂血症、脂肪肝等,结合其临床诊断标准,可诊断为Alstrom综合征。
Alstrom综合征由ALMS1基因突变所致。该基因位于染色体2p13,包含23个外显子,编码4 169个氨基酸。该蛋白广泛表达于中枢神经系统、光感受器、内分泌系统、心肺转流术系统及泌尿生殖系统的纤毛细胞中心体和基底部,在鞭毛运输及维持纤毛细胞功能中起着重要作用[16]。迄今,已报道270种ALMS1基因突变,包括点突变、缺失、插入及框架位移等。大多数发生在外显子8、10和16,其中,外显子16的突变占总突变率的36%[17]。另亦有内含子区域的突变报道[18]。c.10775delc是最常见的突变类型,占了英国病例的50%[9]。该例患儿发生① c.5418delC,p.Y1807Tfs*23,移码变异、杂合;② c.10549C>T;p.Q3517*,无义变异、杂合,分别位于第8和16外显子。其中,c.5418delC为未报道的新变异。序列分析显示该变异导致氨基酸出现移码突变(p.Y1807Tfs*23),蛋白质合成提前终止,产生截断的蛋白。根据美国医学遗传学学会(American College of Medical Genetics,ACMG)的判定标准[19],该变异位点为致病性(pathogenic)变异,其证据包括:(1)非常强烈的证据(pathogenic-very strong,PVS1):移码变异(frameshift);(2)中等证据(pathogenic-moderate, PM2):频率(allele frequency)为0,即在正常人数据库未见该变异位点(包括1000 Genomes,http://www.1000genomes.org/ 和ExAC数据库,http://exac.broadinstitute.org );(3)支持性的证据(pathogenic-supporting):临床表型吻合。
临床上根据其变异位点的不同,可有明显表型变异,如发病时间和严重程度不同[9]。发生于外显子16的突变,临床表型多较严重,表现为心脏、眼、耳、肝脏、肾脏等多器官病变,且有糖类、脂类代谢异常。该患儿临床表现与16号外显子突变相符。另外显子8突变的患者肾脏疾病的发生率较低。但只有一个杂合子突变与有两个杂合子突变的患者相比,其表型表达无明显差异[20]。另研究表明,遗传修饰、环境或感染暴露、随机事件,均可导致AS发病年龄和严重程度发生极大的差异。
Alstrom综合征预后极差。90%患者16岁时完全失明,最终所有患者均会失明。童年期多死于心脏衰竭(约90.5%),而成年期为心脏或肾衰竭致死(61.3%),生存超过50岁的极为罕见[12]。对于该类疾病,目前尚无有效治疗手段,多为对症处理。通过改变生活方式,可以改善代谢综合征。早期佩戴视听矫正器可改善其生活质量。
本例患儿仍在内分泌及心内科门诊随访中,现虽肾功能正常,但超声显示肾脏已有增大,故应考虑其肾脏可能已存在纤维病变。后期应积极监测其肾功能及肺部病变。
鉴于目前Alstrom综合征已具有确诊意义的基因检测方法,早期诊断能评估患儿病情,并可多学科联合治疗。临床对早期视网膜病变,伴心脏疾患、肥胖、耳聋及糖尿病患者,应考虑此疾病可能,及早进行基因检测以明确诊断,有利于患儿尽早进行病情评估,并对其多器官进行性衰竭进行早期综合干预,以期改善预后和长期生存。对有阳性家族史的孕妇则应进行遗传咨询。
[1]Alstrom CH, Hallgren B, Nilsson LB, et al. Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: a specific syndrome (not hitherto described) distinct from the Laurence-MoonBardet-Biedl syndrome: a clinical, endocrinological and genetic examination based on a large pedigree [J]. Acta Psychiatr Neurol Scand Suppl, 1959, 129(1): 1-35.
[2]李梅, 夏维波, 金自孟,等. Alstrom综合征一例 [J]. 中华儿科杂志, 2004, 42(6): 471.
[3]李德天, 王艳秋, 李玲,等. Alstrom综合征一例 [J]. 中华内科杂志, 2004, 43(9): 650.
[4]白然, 王浩, 杨郁, 等. Alstrom综合征一例 [J]. 中华内分泌代谢杂志, 2006, 22(1): 93.
[5]栾云,焦铁健,刘坤. Alstrom综合征1例 [J]. 中国当代儿科杂志, 2007, 9(4): 369G70.
[6]董凤芹, 王保法, 李红, 等. Alstrom综合征一例 [J]. 中华医学杂志, 2007, 87(36): 2592.
[7]匡蕾, 王炜, 叶山东, 等. Alstrom综合征一例 [J]. 中华糖尿病杂志, 2013, 5(3): 189-190.
[8]王丹丹, 倪世宁, 石星, 等. Alstrom综合征患儿ALMS1基因突变分析 [J]. 南京医科大学学报(自然科学版), 2014, 34(10): 1378-1381.
[9]Marshall JD, Hinman EG, Collin GB, et al. Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alstrom syndrome [J]. Hum Mutat, 2007, 28(11): 1114-1123.
[10]Aldahmesh MA, Abu-Safieh L, Khan AO, et al. Allelic heterogeneity in inbred populations: the Saudi experience with Alstrom syndrome as an illustrative example [J]. Am J Med Genet A, 2009, 149A(4): 662-665.
[11]Marshall JD, Muller J, Collin GB, et al. Alström Syndrome: Mutation Spectrum of ALMS1 [J]. Hum Mutat, 2015, 36(7): 660-668.
[12]Marshall JD, Bronson RT, Collin GB, et al. New Alström syndrome phenotypes based on the evaluation of 182 cases [J]. Arch Intern Med, 2005, 165(6): 675-683.
[13]Izzi C, Maffei P, Milan G, et al. The case familial occurrence of retinitis pigmentosa, deafness, and renal involvement [J]. Kidney Int, 2011, 79(6): 691-692.
[14]Manara R, Citton V, Maffei P, et al. Degeneration and plasticity of the optic pathway in Alstrom syndrome [J]. Am J Neuroradiol, 2015, 36(1): 160-165.
[15]Casey J, McGettigan P, Brosnahan D, et al. Atypical Alstrom syndrome with novel ALMS1 mutations precluded by current diagnostic criteria [J]. Eur J Med Genet, 2014, 57(2-3): 55-59.
[16]Collin GB, Marshall JD, Ikeda A, et a1. Mutations in ALMSl cause obesity, type 2 diabetes and neurosensory degeneration in Alstrom syndrome [J]. Nat Genet, 2002, 31(1): 74 -78
[17]Marshall JD, Maffei P, Collin GB, et al. Alström syndrome: genetics and clinical overview [J]. Curr Genomics, 2011,12(3): 225-235.
[18]Bond J, Flintoff K, Higgins J, et al. The importance of seeking ALMS1 mutations in infants with dilated cardiomyopathy [J]. J Med Genet, 2005, 42: e10.
[19]Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [J]. Genet Med, 2015, 17(5): 405-424.
[20]Shenje LT, Andersen P, Halushka MK, et al. Mutations in Alstrom protein impair terminal differentiation of cardiomyocytes [J]. Nat Commun, 2014, 5: 3416.
Alstrom syndrome in children: a case report and literature review
CAI Qingxia1, CHANG Guoying2, DING Yu2, LI Juan2, CHENG Qing2, LI Xin2, WANG Jian2, WANG Xiumin2, SHEN Yiping2
(1. Longyan Hospital of Traditional Chinese Medicine, Longyan 364000, Fujian, China; 2. Shanghai Children’s Medical Center Af fi liated to Shanghai Jiaotong University School of Medicine, Shanghai 200127, China)
ObjectiveTo analyze the clinical feature, diagnosis and treatment of Alstrom syndrome.MethodThe clinical data of a case of Alstrom syndrome and the result of her ALMS1 sequencing by the two generation sequencing were retrospectively reviewed.ResultsThe 12 year and 10 month old female suffered from dilated cardiomyopathy, obesity, optic nerve diseases, sensorineural hearing loss, high blood glucose and irregular menstruation since one month of birth. Laboratory examination showed she had high testosterone level, hyperglycemia, hyperlipidemia and fatty liver. High-throughput sequencing con fi rmed there was ALMS1 gene mutation which includes hybrid frameshift mutations of c.5418delC and p.Y1807Tfs*23, and heterozygous nonsense mutation of c.10549C>T and p.Q3517*, and c.5418delC was a new variation reported for the fi rst time.ConclusionAlstrom syndrome is an autosomal recessive genetic disease, which is characterized by multiple organ dysfunction and metabolic syndrome, and can be diagnosed by gene detection.
alstrom syndrome; ALMS1 gene; gene mutation; genetic counseling
10.3969/j.issn.1000-3606.2017.04.009
2016-09-03)
(本文编辑:邹 强)
王秀敏 电子信箱:wangxiumin1019@126.com
*共同第一作者
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
中国生殖健康(2020年4期)2021-01-18
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
中国临床医学影像杂志(2019年1期)2019-04-25
中国生殖健康(2018年4期)2018-11-06
百科知识(2015年18期)2015-09-10
中国当代医药(2015年31期)2015-03-01
中国医药科学(2015年15期)2015-02-27
湖北农业科学(2014年11期)2014-09-10
