
Potential antioxidant and cytoprotective effects of essential oil extracted from Cymbopogon citratus on OxLDL and H2O2 LDL induced Human Peripheral Blood Mononuclear Cells(PBMC)
2017-05-22 09:01:04JmunSkeenSdullhAshokkumrGokulShnmugnthnSenguttuvnSivnMozhiNirnjliDevrj
食品科学与人类健康(英文) 2017年2期
S.Jmun,Skeen Sdullh M.S.,R.Ashokkumr,Gokul Shnmugnthn,Senguttuvn Sivn Mozhi,Nirnjli Devrj S.,*
a Department of Biochemistry,University of Madras,Guindy Campus,Chennai 600025,India
b Department of Biotechnology,Prist University,Puducherry,India
Abstract Cymbopogon citratus(lemon grass)is commonly used in traditional folk medicine.The essential oil extracted from C.citratus has been reported as a potential anti-oxidant and anti-inflammator agent.This study has been designed to explore the protective effect of C.citratus(lemon grass)againstmodifie LDL(OxLDL and H2O2 LDL)induced cytotoxicity in PeripheralBlood MononuclearCells(PBMC).The essential oilextracted from C.citratus(EOC)was subjected to FT-IR spectroscopic analysis for the identificatio of functionalgroups.In vitro antioxidant assays were carried out to assess the electron donating capability of EOC as compared w ith a known standard L-ascorbic acid.The cytoprotective effects of EOC were determ ined in PBMC induced w ith modifie LDL.Spectra obtained from FT-IR analysis showed the presence of functional groups in EOC such as H-bonded,O-H stretching,N H stretching,aldehyde-C-H stretching,aldehyde/ketone-C-O stretching,-C-C-stretching,-CH3 bending,C-H in plane bending.EOC has greater antioxidant property when compared w ith the standard L-ascorbic acid.EOC at all test concentrations demonstrated free radical scavenging activity and cytoprotective effect when challenged against modifie LDL in PBMC.The above results show EOC as a promising antioxidant and cytoprotective agent.
Keywords:Cymbopogon citratus;Antioxidants;Cytoprotective;PBMC;Modifie LDL
1.Introduction
Cymbopogon citratus(lemon grass),a native herb from India,is also cultivated in other tropical and subtropical countries.It is consumed as an aromatic drink and used in traditional cuisine for its lemon flvour,and is also employed in popular medicine.Infusion or decoctions of dry leaves have been utilized as antim icrobial [1],free radical scavenging,antioxidant[2,3],anti-inflammator [4],antihepatotoxic[5]and antihypertensive agents.In many countries,it is used to treat fever as a relaxant and sleeping aid.Studies onC.citratusleaf extract have demonstrated its anti-inflammator,hypotensive,hypocholesterolem ic [6],vasorelaxing and diuretic effects.Its efficien y against oxidative damage and cancer chemopreventive properties have also been reported by Sharma et al.[7]and Ramar Thangam et al.[8].
The phytochem ical compounds identifie inC.citratusare mainly terpenes,alcohols,ketones,aldehyde and esters.Some of the reported phytoconstituents are essential oils that contain Citralα,Citralβ,Nerol,Geraniol,Citronellal,Terpinolene,Geranyl acetate,Myrecene and Terpinol Methyl heptenone[9,10,11].
Atherosclerosis is define as the loss of elasticity of the arterial wall due to accumulation of LDL beneath the endothelial cells[12].This leads to migration of smooth muscle cells by suppressing NO levels.The current research scenario focuses on the restraint in the modificatio of LDL and subsequent formation of foam cells which impaired lipase activity and accumulation of ROS[13].
In the early phase,the native LDL coming from the liver gets accumulated in the sub-endothelial space of the artery;where they may be minimally oxidized by the enzymes phospholipases,sphingomyelinase and 12/15-lipoxygenase to give rise to products that are atherogenic Levitan et al.[42].Monocytes generally flw in the blood stream and when there is an inflammator response in the artery walls they get differentiated into macrophages.The accumulation of macrophages,chemokines,cytokines and metalloproteinases and continuous presence of modifie lipoproteins result in the formation of complicated atherosclerotic lesions.These lesions are characterized by migration of smooth muscle cells from the middle layer of artery into the sub endothelial layer[14,15].
As suggested by Khalid Rahman[16],the oxidation of LDL begins once the endogenous lipophilic anti-oxidants such as α-tocopherol,α-carotene and ubiquinol levels get completely exhausted.Therefore,additional sources ofinatural antioxidants would be required.Thus,we derived the main objectives of this study as:(1)extraction of essential oil fromC.citratus(2)FT-IR analysis to fin the presence of functional groups in the essential oil(3)in vitroanti-oxidant assays and(4)cytoprotective effect of essential oil fromC.citratusin PBMC induced with modifie LDL(OxLDL and H2O2LDL).
2.Materials and methods
2.1.Essential oil extraction from C.citratus(EOC)
Essential oil ofC.citratus(EOC) was prepared by the reported method of Uzama[17].Briefl,shade dried leaves were powdered and extracted using 70%(v/v)ethanol for 48 h at room temperature.After vacuum filtration the extract was evaporated under vacuum to yield an oily residue.The residue was resuspended and dissolved in sterile water.The aliquots were stored at-20°C and used for further study.
2.2.Fourier transform infrared(FT-IR)spectrometry
FT-IR spectroscopic analysis of EOC was done using Thermo scientifiNicolet IS-5,spectral range of 400-4000 cm-1.EOC samples were prepared by making a pellet in potassium bromide(KBr).Four scans were performed at a resolution of 4.00 cm-1at a scan speed of 0.20 cm/s.Spectral data obtained were plotted on a graph of transmittance(%)versus wave number/cm[18].
2.3.In vitro antioxidant assays
2.3.1.DPPH assay
The antioxidant capacity of the EOC was estimated as reported by Sahoo et al.[43]and compared withL-ascorbic acid(positive control)using the stable DPPH reagent.Different concentrations of the extract (25-100 μg/mL) were dissolved and made upto 1 mL with methanol.1 mL of DPPH reagent was added.1 mL of methanol was then added and the tubes were incubated for 10 min in dark.Standard concentrations of ascorbic acid were also treated in the same way.The absorbances were measured at 517 nm in Shimadzu UV-vis spectrophotometer.The free radical scavenging activities of the extracts were calculated as a percentage of radical reduction.Radical scavenging activity %=[1-(absorbance of control/absorbance of sample)]×100.
2.3.2.Total antioxidant assay
The total antioxidant activity of the EOC was evaluated by the phosphomolybdenum assay as described previously [19].Briefl,0.3 mL of EOC ranging between 25-100 μg/mL was added to 3 mL reagent containing 0.6 mol/L H2SO4,28 mmol/L of sodium phosphate,4 mM of ammonium molybdate and incubated at 95°C for 90 min.The absorbance was measured at 695 nm using a Shimadzu UV-vis spectrophotometer.
2.3.3.Nitric oxide(NO)scavenging activity
Nitric oxide (NO) scavenging activity of the extracts was determined using Griess reaction [20].Sodium nitroprusside(5 mM) in phosphate-buffered saline was mixed with different concentrations of EOC and incubated at 25°C for 150 min.The samples from the above were reacted with Greiss reagent(1%sulphanilamide,2% H3PO4and 1%N-(1-naphthyl) ethylenediamine dihydrochloride).The absorbance of the chromophore formed during the diazotization ofinitrite with sulphanilamide and subsequent coupling with napthylethylenediamine was read at 546 nm and referred to the absorbance of standard solution of Sodium nitrite treated the same way with Griess reagent.
2.3.4.Superoxide anion scavenging assay
The superoxide scavenging activity of the extract was determined by light induced superoxide generation with riboflvin and subsequent reduction ofinitro blue tetrazolium.Different concentrations of EOC ranging from 25 to 100 μg/mL were added to the reaction mixture containing 3 mg of NaCN in 0.1 mol EDTA,0.12 mmol/L riboflvin and 0.6 mol phosphate buffer(pH 7.8)in a fina volume of 3 mL.The tubes containing the reaction mixture were continuously illuminated with incandescent lamp for 15 min.The optical density was read at 530 nm before and after illumination[21].
2.3.5.Reducing power assay
EOC at different concentrations was mixed with phosphate buffer(0.2 mol,pH 6.6)and 30 mM of K3FE(CN)6.The mixture was incubated at 50°C for 20 min.To this,10%TCA was added to stop the reaction and centrifuged at 3000 rpm for 10 min.The upper layer was mixed with equal volume of distilled water and 0.1%FeCl3and the absorbance was measured at 700 nm[22].
2.3.6.Hydrogen peroxide scavenging assay
According to Ruch et al.[23],a solution of hydrogen peroxide(40 mM) was prepared in phosphate buffer (pH 7.4).EOC at different concentrations(25-100 μg/mL)was added to 0.6 mL of H2O2solution.The total volume was made up to 3 mL with phosphate buffer.The absorbance of the reaction mixture was recorded at 230 nm against the blank solution which contained phosphate buffer without H2O2.
2.3.7.Anti-hemolytic activity
Human whole blood was washed three times in 9 volumes of sterile,0.9% saline solution.Different concentrations of EOC and oxidative stress reagent (H2O2) were added to the isolated erythrocytes.100 μL of 5% erythrocytes were diluted in PBS and 50 μL of EOC containing different concentrations(25-100 μg/mL PBS,pH 7.4) were added.The reaction mixture was diluted with 3 mL of PBS and centrifuged at 2000×gfor 10 min.The absorbance from the resultant supernatant was read at 540 nm[24].
2.4.Isolation of LDL+VLDL from human plasma
When 2 g of sucrose was added to 2 mL of plasma,the volume increased to 3.2 mL.
To this solution,32 μL of 5%heparin and 160 μL of 2 mol/L MgCl2were added.LDL+VLDL precipitated immediately.After 15 min at room temperature,the mixture was centrifuged in stoppered tubes at 6000×gfor 30 min.The high density of the sucrose solution causes the precipitated lipoproteins to floa to the top and form a pellicle on the surface of the clear supernatant.The supernatant was removed.The tubes were centrifugedfor1 mintosedimentthepellicleandthe,precipitatewas dissolvedin40 μLof5%NaClbyincubationfor30 minat37°C.Lipoprotein was precipitated again by adding 2 mL Tris-HCL buffer and 50 μL of 2 mol MgCl2to remove contaminating serum proteins.The precipitate sedimented upon centrifugation(10 min,6000×g) and was dissolved in 40 μL of 5% NaCl.The washed precipitate was then dissolved in 10%sodium citrate and dialysed against Tris-HCl-NaCl buffer for 24 h at 4°C.After 24 h,the dialysate was centrifuged in cold condition with heparin-barium salt.The precipitate was again dialyzed against Tris-HCl-NaCl buffer to remove the barium ions.The resultant clear yellow solution oflipoprotein was LDL+VLDL[25].The obtained lipoproteins were used for further study.
2.4.1.Preparation of oxidized LDL
Copper sulphate and H2O2were used as catalysts for oxidation based on the method of Witting et al.[26]and Lopes-Virella et al.[27].Briefl,5.0 mL of CuSO4and 30 mmol/L of H2O2were added to 1 mL of LDL+VLDL(200 mg/mL)followed by incubation at 37°C for 24 h and 3 h in dark.At the end of the incubation period,the oxidation of sample was stopped by the addition of 1 mL of 10%TCA.
2.5.Cytotoxicity assay
2.5.1.Isolation of Peripheral Blood Mononuclear Cell(PBMC)
The ficol density gradient separation of whole blood is the most commonly used procedure for separation of mononuclear cells[28].Briefl,0.5 mL of heparin was added to 5 mL of blood in a falcon tube and mixed.To 2.5 mL of ficoll 1 mL of PBS was added in a sterile falcon tube.2.5 mL of blood was layered onto the ficol slowly and centrifuged at 1700 rpm for 20 min.The buffy coat was isolated,completely washed with PBS and centrifuged.Cell viability test was assessed by trypan blue method[29].The cell count was done by counting of cells in haematocytometer.The isolated monocytes were maintained as suspension culture under 5%CO2incubator at 37°C,for further study.
2.5.2.MTT assay
Cells were seeded in a 96 wells plate,at 1×106cells/well and incubated overnight at 37°C and 5% CO2.Complete medium was then replaced with 100 μL incomplete medium containing different concentrations of the drug dissolved in sterile water and incubated for 24 and 48 h,respectively.After incubation,the medium was discarded,100 μL ofincomplete medium and 10 μL of MTT were added in each well.The plates were incubatedfor4 hindark.Afterincubation,themediumwasdiscarded and 100 μL of DMSO was added.The absorbance was measured at 630 and 492 nm in a plate reader[30].
2.5.3.Experimental grouping
To assess the oxidative stress induced by OxLDL and H2O2LDL and the cytoprotective effect of EOC on PBMC,the cells were grouped as:
Control:PBMC (5×106cells) were maintained for 48 h under 5%CO2at 37°C.
Induced:PBMC (5×106cells) were induced with OxLDL(10 μg/mL)/H2O2LDL(5 μg/mL)for 48 h for monocyte/macrophage differentiation.
Treated:PBMC (5×106cells) were induced with OxLDL(10 μg/mL)/H2O2LDL(5 μg/mL)and co-treated with 100 μg/mL of EOC for 48 h.
After 48 h,cells were harvested and used for further studies.
2.5.4.Cytotoxicity assay
2.5.4.1.Trypan blue dye exclusion method.According to Strober[29],Trypan blue(0.4%solution)staining was used to document the viability of suspension and adherent PBMC with 100 μg/mL of EOC.After 48 h at 37°C,the adherent PBMC were washed by centrifugation and fresh medium was added.10 μl of cells from each experimental group were stained with trypan blue dye (0.4% w/v) and the numbers ofiviable and dead cells were scored under a light microscope with Neubauer hemocytometer.
2.5.4.2.LDH leakage assay.To evaluate the potential cytotoxic effect of OxLDL and H2O2LDL on PBMC,lactate dehydrogenase (LDH) released in the supernatant was determined as described by King (1965).The percentage of cellular LDH release was calculated as the portion of LDH detected in the conditioned medium divided by total LDH from conditioned medium plus LDH from the cell lysate.
2.5.4.3.Quantification ofinitric oxide.Nitrite production,an indicator of NO production,was measured in the conditioned culture medium by the Griess reaction Schulz et al.(1999).The supernatants were collected for the quantificatio ofinitrite.Briefl,100 μl of supernatants were mixed with 100 μl of Griess reagent(equalvolumesof1%(w/v)sulphanilicacidin30%(v/v)acetic acid and 0.1% (w/v)N-(1-naphthyl) ethylenediamine in 60% (v/v) acetic acid) and incubated at room temperature for 30 min in the dark.The absorbance was measured at 540 nm spectrophotometrically.
2.5.4.4.Quantification ofintracellular ROS generation.Intracellular ROS generation by PBMC was assessed by the nitroblue tetrazolium(NBT)reductionassayRooketal.(1985).Thisassay is based on the ability of phagocytic cells to produce superoxide upon stimulation with OxLDL and H2O2LDL.Cell suspension was incubated with an equal volume of 1% NBT dissolved in PBS at 37°C,for 30 min in the dark.After the incubation time,it was examined under a light microscope for blue-black nitroblue diformazan deposits,indicative of OxLDL and H2O2LDL stimulated respiratory burst.At least 200 cells were assessed for each experiment.After that,blue-black nitroblue diformazan was solubilized with 2 mol KOH and DMSO and the absorbance at 630 nm was measured spectrophotometrically.A standard curve was prepared by adding KOH and DMSO to a known concentration of NBT and 100 μM H2O2was added to the cells as positive control.
2.5.4.5.Lipid peroxidation.The TBARS assay is a modifie method of Buege and Aust[31].0.5 mL oflysates were mixed thoroughly with 1 mL of a solution containing 15% (w/v)trichloroacetic acid,0.375% (w/v) TBA,and 0.25 N HCl,and heated for 15 min.After cooling,the flocculen precipitates were removed by centrifugation.The absorbance of the sample was determined at 535 nm against a blank containing all reagents except samples.The level oflipid peroxides is expressed as ng of MDA released/mg protein.
2.5.5.Statistical analysis
All the experiments were carried out in triplicate and results are given as the mean±standard deviation.The data were analyzed (Microsoft Excel 2007) for statistical significanc using Studentst-test and differences were considered significan at p <0.05.
3.Results
3.1.FT-IR spectrum of essential oil extracted from C.citratus(EOC)
The infrared spectra of the EOC were recorded by Thermo scientifiNicolet IS-5 and run under infrared region of 400-4000 cm-1range.Stretching of-CH3was observed corresponding to an alkyl saturated aliphatic group and at 2925.75 and 2854 cm-1symmetric and asymmetric stretching of-CH2were observed.The intense band observed at 1724.83 cm-1is due to vibrations of C=C (cisandtrans),confirmin the presence of conjugated double bonds (C=C-CHO) in citral which are common in acyclic monoterpenes.The peak at 1629 cm-1indicated the stretching of C=O of the aldehyde group.At the 1383.65 cm-1peak,bending of the-CH2group was observed.At 1229.02 cm-1bending of-CH3group was observed.From 1194 to 1048.64 cm-1,stretching of-C-O and vibrations of the-CH skeleton were observed in Fig.1 and Table1.Similar peaks were reported by Wany et al.[18]for Geranial from another species ofCymbopogon.
3.2.Antioxidant potential of essential oil extracted from C.citratus
The scavenging activity of the EOC against DPPH was concentration-dependent (Table2).The highest concentration(100 μg/mL)of EOC showed highest DPPH scavenging activity when compared to the other concentrations of EOC.Interestingly,EOC was as effective as the positive control,L-ascorbic acid,in scavenging 50%of DPPH radicals present in the reaction mixture.
EOC was found to possess significan reducing power and antioxidant activity (Table2).100 μg/mL concentration was found to be more active when compared to the lower concentrations.Reducing power of EOC and at antioxidant activity 100 μg/mLwerecomparablewithstandardL-ascorbicacid.This was due to the presence ofivarious phytochemicals in higher proportions.
EOC was found to exhibit better inhibition of superoxide radical (Table2) at concentrations of 75 μg/mL and 100 μg/mL.The lower concentration (25 μg/mL) did not show much inhibition when compared to higher concentrations.100 μg/mL of EOC showed comparable inhibition of superoxide radical withL-ascorbic acid.
H2O2scavenging activity was evaluated by a decrease in the formation of chromogen in the Fenton reaction (Table2).The H2O2scavenging activity was highest at 100 μg/mL,when compared withL-ascorbic acid.The percentage NO inhibition by various concentrations of EOC (100 μg/mL,75 μg/ml,50 μg/mL and 25 μg/mL)respectively was found to be 62%,50%,35%and 16%.From the obtained results,100 μg/mL and 75 μg/mL of EOC showed 50% inhibition ofinitric oxide,whereas the lower concentration(25 μg/mL)showed only 16%ofinhibition.Higher concentration (100 μg/mL) was found to be effective in nitric oxide scavenging compared withL-ascorbic acid.
The results obtained show the inhibition of hemolysis at each concentration of the EOC.As shown in Fig.2,the protective effect of EOC against hemolysis was directly proportional to the concentration of the extract.The degree of protection was of the order 100 >75 >50 >25 μg/mL.However,the level of protection afforded by the extracts was less than that of L-ascorbic acid.

Fig.1.FT-IR spectrum of essential oil of Cymbopogon citratus.Region I may correspond to the presence of cycloalkanes,region II to the presence of terpenoids and flvonoids and region III to that of methyl group.
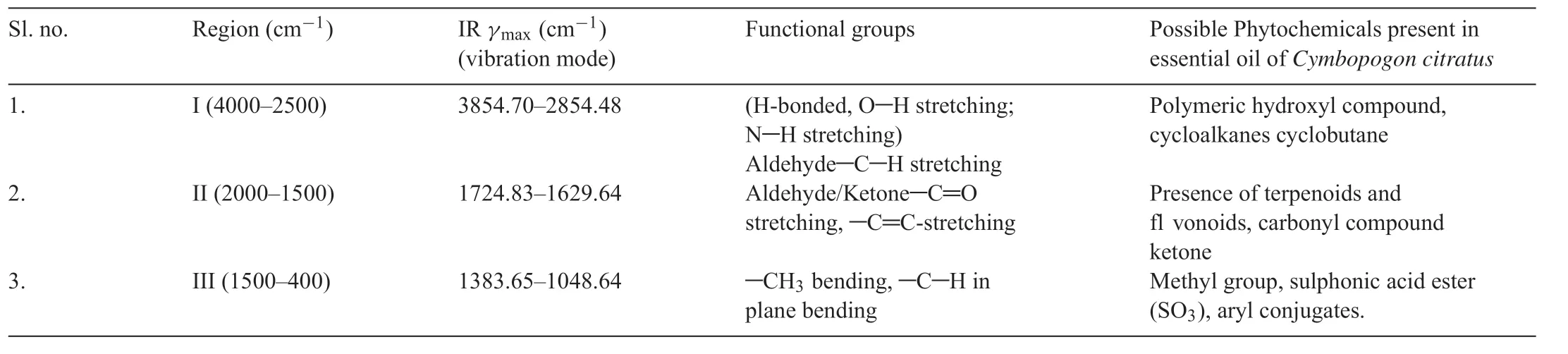
Table1 FT-IR spectra of essential oil of Cymbopogon citratus.
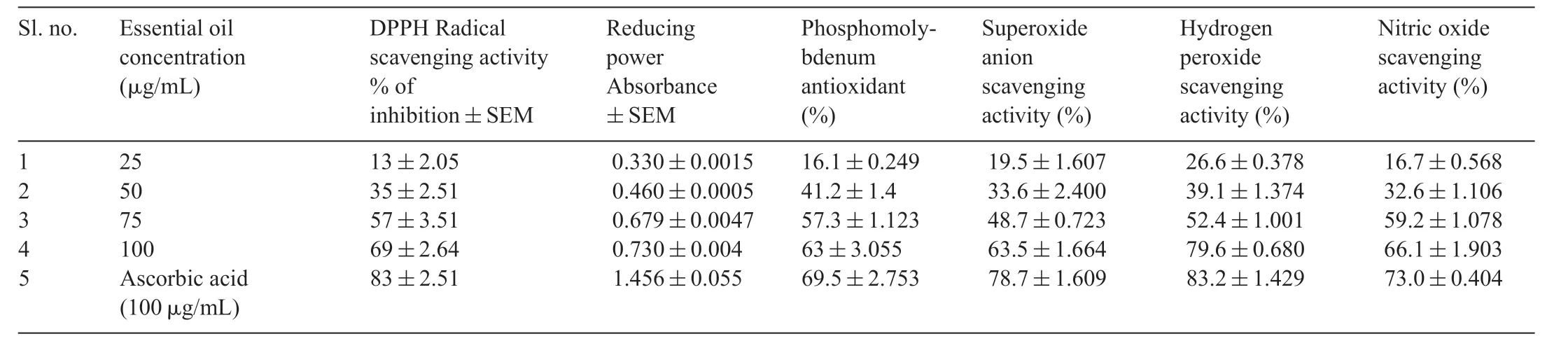
Table2 In vitro antioxidant potential of essential oil extracted from Cymbopogon citratus.

Fig.2.The percentage ofinhibition of hemolysis by EOC at different concentration (25-100 μg/mL) compared to L-ascorbic acid (100 μg/mL) as standard.Experiment was performed in triplicate and data shows the mean±SD(n=3).
3.3.Cytoprotective effect of essential oil extracted from C.citratus
The percentage of PBMC viability was tested for different concentrations of EOC,OxLDL and H2O2LDL.As shown in Fig.3(A and B),there was no cytotoxicity at any of the concentrations of EOC.Hence we found 100 μg/mL of EOC was a suitable concentration for further study because of the presence of phytochemicals at higher concentration.PBMC incubated with OxLDL and H2O2LDL concentrations ranging between 5-25 (μg/mL of protein) showed maximum viability at 10 μg/mL for OxLDL and 5 μg/mL for H2O2LDL.
As shown in Fig.4(A) the cytoprotective effect of EOC on PBMC induced with OxLDL and H2O2LDL for 48 h,showed cellular toxicity by decreasing cell viability which was assessed by trypan blue exclusion assay.EOC exhibits cytoprotection against OxLDL and H2O2LDL as observed by the significant reduction in cell death.We assessed cellular integrity using LDH leakage assay as depicted in Fig.4(B).The PBMC treated with various concentrations of EOC showed significan reduction in LDH leakage.Therefore,results from MTT,trypan blue exclusion assay and LDH leakage assay can be correlated and concluded that EOC could efficientl reduce the cytotoxicity produced by oxidative stress which was induced by modifie LDL(H2O2LDL and OxLDL).
PBMC treated with EOC induced with OxLDL and H2O2LDL demonstrated significan reduction in ROS generation(Fig.5A).The effect ofC.citratusextract on the production of the proinflammator mediator NO was analysed by measuring the nitrite released into the medium.Fig.5(B) shows increased NO production in the OxLDL and H2O2LDL induced PBMC cells when compared to the EOC treated groups.The above results suggest the potent antioxidant and cytoprotective activity ofC.citratus.The inhibition oflipid peroxidation by the essential oil ofC.citratuson PBMC cells induced with OxLDL and H2O2LDL was measured by the color intensity of the MDA-TBA complex.The result demonstrates inhibition oflipid peroxidation by EOC(Fig.6).
4.Discussion
The utilization of medicinal plants has relied largely on longterm clinical experience with little or no scientifidata on their effica y or safety after long-term treatment (Zhu et al.,2002;Ernst,2004).As is the case in relation to many other medicinal plant species,there are few toxicological/toxicogenetic studies on the safety ofC.citratus.
Degenerative disorders,such as cancer,cardiovascular diseases and neurodegenerative diseases progress due to high levels of ROS or free radicals Yen et al.(2002).Plant based antioxidants can elicit increased synthesis of endogenous antioxidant defenses or by themselves act directly as antioxidants[32].
The cardioprotective effect ofC.citratusin isoproterenolinduced cardiotoxicity has been reported by Gayathri et al.(2010) who showed that lemon grass administration decreases the toxic effects oflipid peroxidation in both serum and heart tissue by increasing the levels of antioxidants.Costa et al.(2011)showed that oral intake ofC.citratus essentialoil for 21 days showed reduction in cholesterol and genotoxicity.
DPPH radical scavenging ability of EOC was comparable with standardL-ascorbic acid suggesting the presence of proton donating ability of EOC that could serve as free radical inhibitor.The reducing power of EOC suggested that the strong antioxidant activity of EOC might be due to the presence of phenolic compounds which absorb and neutralize free radicals and quench peroxides,due to their redox properties(Gordan,1990;Osawa et al.,1994).
The total antioxidant capacity of EOC was assessed by phosphomolybdenum method.Luximon-Ramma et al.(2005).In the present study,the obtained results demonstrate that the highest antioxidant activity was at the highest concentration of EOC used(100 μg/mL).The difference in the antioxidant capacity of EOC is because of the presence of phytochemicals such as tannins,triterpenoids,steroids and flvonoids[33,34].Our results indicate that EOC has good antioxidant potential.
Superoxide and hydroxyl radical are the two major ROS produced during the reduction of oxygen to water.Superoxide radicals are formed by one electron reduction of molecular oxygen [35,36].Superoxide scavenging activity of EOC was comparable with the known antioxidantL-ascorbic acid,thus EOC also possesses superoxide scavenging activity.

Fig.3.Effect of the EOC(A)and(B)OxLDL and H2O2 LDL on cell viability of PBMC assessed by MTT assay.Experiment was performed in triplicate and data shows the mean±SD(n=3).
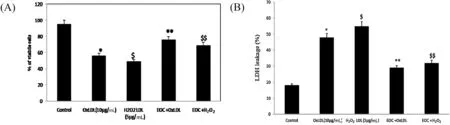
Fig.4.Cytotoxic effect of OxLDL and H2O2 LDL.(A) Trypan blue exclusion test (B) LDH leakage assay *p <0.05 compared to control group;$p <0.05 in comparison with the OxLDL induced group.**p <0.05 compared to control group;$$p <0.05 in comparison with the H2O2 LDL induced group.
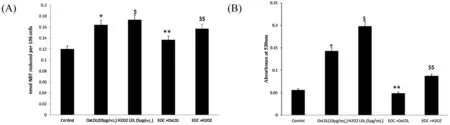
Fig.5.Inhibition of ROS(A)and nitric oxide production(B)in PBMC induced with OxLDL and H2O2 LDL and treated with EOC.*p <0.05 compared to control group;$p <0.05 in comparison with the OxLDL induced group.**p <0.05 compared to control group;$$p <0.05 in comparison with the H2O2 LDL induced group.
Nitric oxide can be described as a pleiotropic inhibitor of physiological process because it plays a vital role in smooth muscle relaxation,neural signalling,inhibition of platelet aggregation and regulation of cell mediated toxicity[37].Our results exhibit that EOC could increase the demand for NO under oxidative stress and since heart diseases can occur in a background of oxidative stress,the EOC may be considered to contain cardioprotective property.
Leeetal.[38]havestatedthatH2O2ispoorlyreactivebecause ofits weaker oxidizing and reducing capabilities.Hence,biologically it can be a cytotoxic agent by converting itselfinto hydroxyl radical in the presence of metal ions and superoxide anion and produces singlet oxygen through reaction with superoxide anion or with hypochlorous acid.In this study,EOC was less effective at lower concentration,whereas at 100 μg/mL the H2O2scavenging activity was more as compared toL-ascorbic acid.
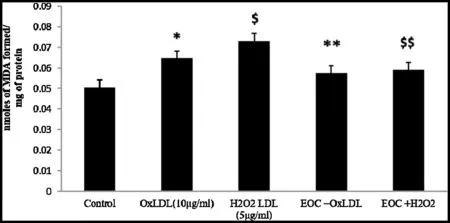
Fig.6.Formation of MDA indicates the lipid peroxide levels in PBMC induced with OxLDL and H2O2 LDL and treated with EOC *p <0.05 compared to control group;$p <0.05 in comparison with the OxLDL induced group.**p <0.05 compared to control group;$$p <0.05 in comparison with the H2O2 LDL induced group.
H2O2can degrade haemoglobin,to release Fe2+ions that can lead to erythrolysis.Azizova et al.[39]showed that erythrocyte membranes during atherosclerotic conditions are prone to lysis in the presence of oxidized LDL.We found that EOC could efficientl inhibit lipid peroxidation,which supports its potential to stabilize erythrocyte membranes thereby acts as an anti-inflammator agent.The erythrocyte membrane resembles the lysosomal membrane and,as such,the effect of drugs on the stabilization of erythrocyte membrane could be extrapolated to the stabilization oflysosomal membrane [44].Thus,EOC exerts its protective effect on the lysosomal membrane which could otherwise lead to oxidative stress induced cell death.
OxLDL is the product of oxidation of their polyunsaturated fatty acid component due to the increased concentration of ROS at the subendothelial level.Two aldehydes with strong immunogenic properties are formed:malondialdehyde and 4-hydroxynonenal with anionic valence.Uptake of oxidized LDL renders the macrophages less mobile,thereby promoting the accumulation of these lipid-laden foam cells in the intima.The foam cells retain their metabolic activity and secrete a variety of cytokines and inflammator mediators.The outcome of their activation include recruitment and proliferation of smooth muscle cells which in turn elaborate additional locally active cytokines.Subsequent LDL oxidation causes recruitment of additional monocyte/foam cells and further impairment of endothelial function.
OxLDL and H2O2LDL induction lead to macrophage conversion and glycation of collagen which enhances the entrapment of LDL and its subsequent oxidative modification Thus,there is a release of reactive oxygen intermediates associated with the formation.The increased LDH leakage induced by OxLDL and H2O2LDL cytotoxicity increased the permeability of the cell.Arterial smooth muscle cells modify LDL so that macrophages will take it up faster.The mechanism of LDL modificatio by cells involves lipid peroxidation.Lipid peroxidation was found to be increased upon OxLDL and H2O2LDL induction.On treatment withC.citratusthe levels were significantl reduced.This proves thatC.citratuspossesses strong anti-lipid peroxidation property[45].
The polarity features of the phytochemical components is a key factor that confers their solubility and ability to access the lipid phase where the lipid peroxidation process is taking place,thus influencin their capacity to break chain reactions.Antioxidant compounds with high partition coefficient are preferentially distributed to hydrophobic compartments,thus protecting lipids from free radical attack.
Peroxidation oflipid is probably one of the important intermediary events in oxidative stress-induced cellular damage through the perturbation of cellular redox balance.It has been reported that lipid peroxidation is a persistent process in cells,maintained at basal level,by protective enzymes and antioxidants which prevent it from entering the autocatalytic phase[40].
Inhibition of oxidation of LDL due to the antioxidant potential of the EOC could prevent lipid accumulation within macrophage which in turn could inhibit atherosclerosis.Hishikawa et al.[41]suggested that the antioxidant activities of many polyphenols are responsible for their protection against atherosclerosis.
Phagocytes are crucial in the host defence against invading microorganisms through reactive oxygen species (ROS)production.Measurement of ROS production by phagocytes is of critical importance to investigate the physiological consequences resulting from cellular mechanisms that lead to oxidative burst.ROS production occurs when macrophages were stimulated by modifie LDL.
The NBT reduction assay demonstrates the production of superoxide anion by accepting electrons from a variety of donor substances,such as superoxide,and thus is converted into a reduced form which precipitates as a blue-black insoluble material (formazan) in the cytoplasm of phagocytes.Reduced respiratory burst activity when treated with EOC could be due to the blocking of potassium channels of macrophages,as described.
Figueirinha et al.[4],suggested that the anti-inflammator activity ofC.citratusextract is due to its inhibition of NO production and iNOS expression in dendritic cells.Nitric oxide production was independent of the concentration of exogenousL-arginine.In the present study,nitric oxide production and respiratory burst activity were inhibited by EOC.This supports the subsequent depolarization of the plasma membrane,which may lead to the impairment ofintracellular signalling steps required for macrophage activation.
To conclude,our findin has excavated knowledge on drug formulation ofC.citratuswhich exhibits negligible cytotoxic effect on normal cells and demonstrates antioxidant properties as evidenced from in vitro antioxidant assessment,has anti-inflammator properties as evidenced by the inhibition of hemolysis of erythrocytes and exhibited cytoprotective properties,when added to PBMC induced with OxLDL and H2O2LDL.Thus,EOC showed differential response against deleterious action of ROS,suggesting that this can be validated as a therapeutic and preventive agent against coronary artery diseases.

- 食品科学与人类健康(英文)的其它文章
- About the Beijing Academy of Food Sciences
- α-Glucosidase inhibitor produced by an endophytic fungus,Xylariaceae sp.QGS 01 from Quercus gilva Blume
- Dietary fenugreek(Trigonella foenum-graecum)seeds and garlic(Allium sativum)alleviates oxidative stress in experimental myocardial infarction
- Extraordinary tunable dynam ic range of electrochem ical aptasensor for accurate detection of ochratoxin A in food samples
- PCR-based methodologies for detection and characterization of Listeria monocytogenes and Listeria ivanovii in foods and environmental sources
- GUIDE FOR AUTHORS