
天然磁铁矿和商用Fe3O4修复Hg(II)污染地下水的模拟研究①
2017-04-24 09:36殷鹏华胡志豪
土壤 2017年1期
林 娜,宋 昕,郭 亮,殷鹏华,胡志豪
(1 中国科学院土壤环境与污染修复重点实验室(南京土壤研究所),南京 210008;2 中国科学院大学,北京 100049;3 南京康地环保科技有限公司,南京 210002;4 南京大学现代工程与应用科学学院,南京 210093)
天然磁铁矿和商用Fe3O4修复Hg(II)污染地下水的模拟研究①
林 娜1,2,宋 昕1*,郭 亮3,殷鹏华4,胡志豪4
(1 中国科学院土壤环境与污染修复重点实验室(南京土壤研究所),南京 210008;2 中国科学院大学,北京 100049;3 南京康地环保科技有限公司,南京 210002;4 南京大学现代工程与应用科学学院,南京 210093)
目前汞污染地下水修复面临很大的技术和成本挑战,亟需发展修复效果好、经济效益性高的汞污染地下水修复技术和修复材料。通过批量实验和光谱分析探究了天然磁铁矿和商用Fe3O4对Hg(II) 的去除效率和去除机制,并分析了两种材料对模拟地下水中Hg(II) 的吸附和脱附行为。结果表明,天然磁铁矿和商用 Fe3O4对Hg(II) 的去除受pH、Hg(II) 初始浓度、Cl–等因素的影响;二者对Hg(II) 的去除均符合准二级动力学模型和Freundlich模型;天然磁铁矿对Hg(II) 的去除机制主要是羟基络合与物理吸附,而商用Fe3O4对Hg(II) 的去除主要是化学还原与物理吸附。二者对模拟地下水中Hg(II) 的去除率分别达90% 和95%,具有修复Hg(II) 污染地下水的应用潜力。
天然磁铁矿;商用Fe3O4;修复;Hg(II);地下水污染
汞是环境中毒性最强的重金属污染物之一,广泛存在于世界各地的各种环境介质中,美、欧、亚、非各大洲都存在汞污染问题,甚至在北极地区也受到了汞污染的威胁[1]。地下环境中汞的人为来源包括采矿[2–4]、氯碱生产[5–6]、木材防腐[7]、核武器制造[8]等。人为排放的汞,尤其是溶解度较高的Hg(II),在特定条件下可被转化为毒性更强的有机汞,对人体健康和生态环境造成了严重威胁[9–10]。活性炭[11–13]、介孔SiO2[14]和改性纳米材料[15–18]等已被用于水相溶液中Hg(II) 的去除。Yardim 等[12]利用糠醛活性炭吸附Hg(II),活性炭表面的含氧官能团可与Hg(II) 发生络合吸附。由于巯基对Hg(II)具有很强的亲和力,利用巯基改性介孔 SiO2或纳米 Fe3O4均能够有效地吸附溶液中的Hg(II)[14,16]。然而由于上述材料中大部分材料价格昂贵或制备过程复杂,限制了在地下水Hg(II)污染修复中的应用[10]。
磁铁矿(一般化学式为 Fe3O4)是一种混合价态的铁氧化物,含有Fe(II) 和Fe(III)[19]。对于磁铁矿应用于水相溶液中Hg(II) 的去除已有一些相关研究[10,16,19–21],如成翠兰等[21]的研究认为纳米 Fe3O4对 Hg(II) 的去除机制主要是物理吸附;Wiatrowski等[19]将合成纳米磁铁矿作为还原剂用于还原Hg(II),通过厌氧条件下的 Hg(II) 还原实验和光谱学手段验证了磁铁矿中的Fe(II) 可将Hg(II) 还原为Hg(0)。上述研究表明磁铁矿具有去除Hg(II) 的潜力,但是地下水中Hg(II) 的去除受到极少关注[5,22]。此外,以往关于磁铁矿的研究大多是纯度高、成分均一的合成磁铁矿(主要是纳米磁铁矿[Fe3O4])或精矿[20,23],尚未发现有研究用天然磁铁矿去除地下水中的Hg(II),去除机制也未见明确报道。作为一种天然材料,天然磁铁矿尤其是铁含量低的矿渣十分廉价易得,是大规模地下水修复工程应用的理想材料,具有良好的应用前景[24–26]。可渗透反应墙(permeable reactive barrier,PRB)技术是一种原位地下水修复技术,在欧美已得到广泛的工程应用,针对特定污染物选择有效、经济、安全的活性填充材料是PRB技术成功的关键之一。
本研究首先在去离子水体系中探究不同实验条件(即溶液初始pH、天然磁铁矿用量、反应时间、Hg[II]初始浓度和共存离子)下天然磁铁矿对Hg(II) 的去除效率,研究Hg(II) 去除的影响因素。同时,商用Fe3O4粉末也设置同样的实验与天然磁铁矿的效果进行比较。然后进行了动力学和等温线研究和光谱学分析,探究两种材料对 Hg(II) 的去除机制。最后在模拟地下水体系中通过吸附和脱附实验,分析了两种材料对Hg(II) 污染模拟地下水的修复效率和在地下水中的稳定性,并筛选出合适的PRB填充材料修复Hg(II)污染地下水。
1 材料与方法
1.1 实验材料与试剂
本研究采用了两种修复材料:天然磁铁矿和商用Fe3O4。天然磁铁矿购自河南省巩义市元亨净水材料厂,实验前用自来水和去离子水搅拌、冲洗多次,洗后pH显中性,于烘箱中50℃干燥后过筛得到0.5 ~ 1.0 mm粒径的天然磁铁矿样品备用。实验所用商用Fe3O4(化学纯)购自国药集团化学试剂有限公司,通过扫描电镜图像可判断粒径约为0.5 ~ 20.0 μm。选择两种粒径差异较大的材料目的是探究其在不同含水层中的适用性。两种材料的来源、价格、性状、粒径和预处理方式见表1。
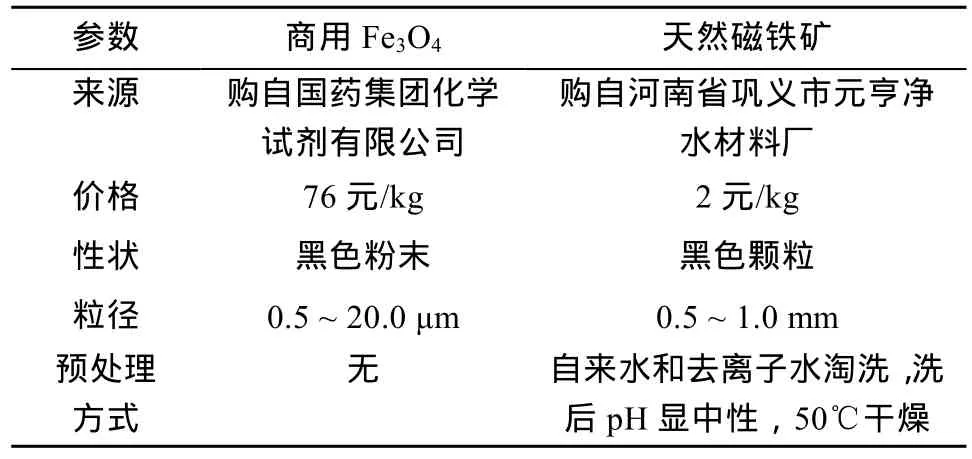
表1 商用Fe3O4和天然磁铁矿比较Table 1 Comparison of natural magnetite and commercial Fe3O4
100 mg/L的 Hg(II) 标准储备液由 Hg(NO3)2×1/ 2H2O用5% 硝酸溶液配制而成,实验所需浓度的含Hg(II) 溶液由该储备液稀释得到。实验用水为去离子水,参考Murphy等[27]使用的无机盐溶液成分,配制本实验所用模拟地下水,各成分最终浓度为:0.1 mg/L FeSO4·7H2O;2 mg/L MgSO4·7H2O;3 mg/L NH4Cl;0.6 mg/L NaH2PO4·H2O;5 μg/L MnCl2、H3BO3、Na2MoO4·2H2O、CoCl2·6H2O、NiSO4·6H2O、CaSO4·5H2O、ZnSO4·7H2O;10 mmol/L哌嗪-1,4-二乙磺酸(PIPES)缓冲溶液,pH = 7.0。模拟地下水中各阴阳离子浓度见表2。除特别说明,所用试剂均为分析纯。
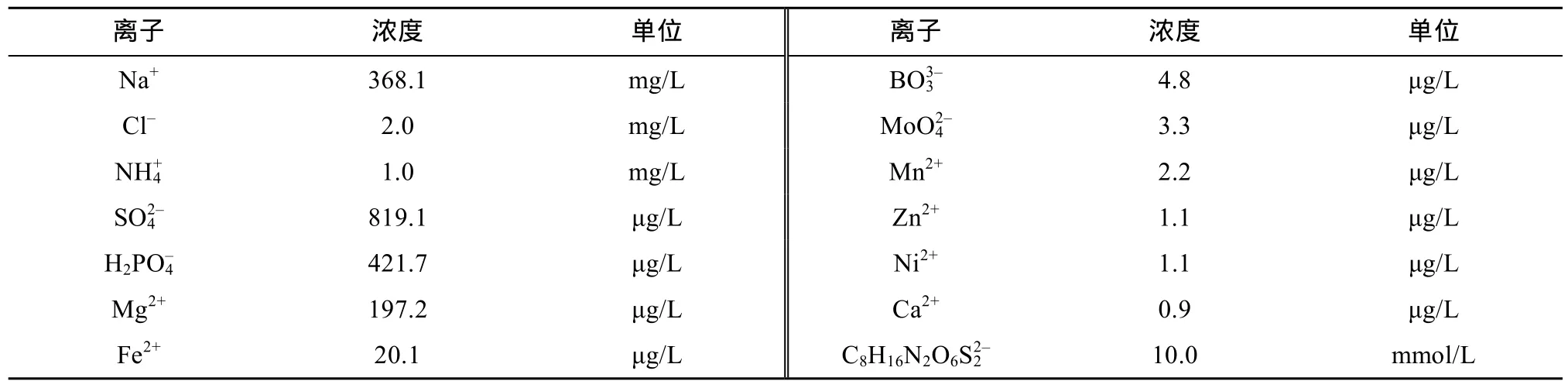
表2 模拟地下水中各阴阳离子浓度Table 2 Concentrations of anions and cations in simulated groundwater
1.2 材料表征和化学成分分析
通过扫描电镜(scanning electron microscope,SEM)(S-3400N II,日本Hitachi)分析商用Fe3O4粒径和形貌;X射线荧光光谱(X-ray fluorescence spectrometer,XRF)(ARL-9800,瑞士ARL)分析天然磁铁矿的化学组成,并用化学滴定确定其中Fe(II) 和Fe(III) 含量;傅里叶变换红外光谱(fourier transform infrared spectroscopy,FTIR)(NEXUS 870,美国NICOLET),判定Hg(II) 吸附前后天然磁铁矿和商用Fe3O4表面基团的变化;X射线光电子能谱(X-ray photoelectron spectroscopy,XPS)(PHI 5000 VersaProbe,日本UlVAC-PHI),分析吸附前后天然磁铁矿和商用Fe3O4表面Fe的化学态变化。
1.3 批量实验
吸附实验:取一定量水洗后粒径为0.5 ~ 1.0 mm的天然磁铁矿或商用Fe3O4粉末分别加入到40 ml棕色硼硅酸盐玻璃瓶中,加入 30 ml浓度为 50 ~ 2 000 μg/L的含Hg(II) 溶液,用0.1 mol/L HNO3和0.1 mol/L NaOH调节溶液到指定的pH,于往复式振荡器中低速振荡一定时间(因振荡器型号限制,无法设置具体振荡转数)。对照样品不添加天然磁铁矿或商用Fe3O4。反应完毕后立即用0.45 μm孔径注射式过滤器过滤,测定滤液中的Hg浓度。每个实验设置3个平行,结果取其均值。实验中仅控制反应的初始条件,除特别说明外,所有实验和样品采集、分析均在室温下进行。
脱附实验:先将一定量的天然磁铁矿和商用Fe3O4置于30 ml Hg(II) 污染模拟地下水中进行吸附实验,然后将其中的固体过滤分离,并用去离子水洗去表面残留的含 Hg(II) 溶液。将吸附 Hg(II)的固体重新加入到30 ml不含Hg(II) 的模拟地下水中振荡60 h后测定溶液中的Hg(II) 浓度。
1.4 分析方法
Hg浓度测定参照《水质 汞、砷、硒、铋和锑的测定 原子荧光法》(HJ 694-2014)[28],所用仪器为原子荧光光度计(AFS-930,北京吉天仪器有限公司),载流为5% HCl。方法检出限为0.04 μg/L,测定下限为0.16 μg/L。
2 结果与讨论
2.1 材料表面形态与化学成分分析
商用Fe3O4的SEM图像如图1所示,可以看出商用Fe3O4表面光滑,形状不规则,粒径约为0.5 ~ 20.0 μm。由商用 Fe3O4的质检信息得知主成分含量≥98%,其中FeO含量为27.0% ~ 29.0%,由此可计算出Fe(II) 和Fe(III) 含量分别为21.0% ~ 22.5% 和48.3% ~ 49.7%,因此其化学计量数x = Fe(II)/Fe(III)为0.42 ~ 0.47。由XRF测定的天然磁铁矿的化学成分见表 3(结果以氧化物的形式表示)。由表可知天然磁铁矿的成分较为复杂,主要元素成分包括Si、Fe、Ca、Mg和Al等。值得注意的是Mn、Cr等有毒元素的含量极低,这一特征有利于降低天然磁铁矿用于地下水修复时造成二次污染的可能性。化学滴定结果表明天然磁铁矿中Fe(II) 和Fe(III) 含量分别为4.9%和 12.5%,通过计算可知其化学计量数 x = Fe(II)/ Fe(III) 为0.39,总Fe含量为17.5%。滴定分析得到的总Fe含量与XRF分析得到的Fe2O3含量为26.5% (即总Fe含量为18.5%)基本一致。

图1 商用Fe3O4的SEM图像Fig. 1 SEM image of commercial Fe3O4

表3 天然磁铁矿的化学组成(%)Table 3 Chemical composition of natural magnetite analyzed by XRF
商用Fe3O4和天然磁铁矿中均含有一定量的Fe(II),且化学计量数均介于0.29 ~ 0.50,根据Pasakarnis等[29]的研究,说明二者均具有一定的 Hg(II) 还原能力。但是商用Fe3O4中的Fe(II) 含量和化学计量数均较天然磁铁矿高,说明商用 Fe3O4更具有还原 Hg(II) 的潜力。
2.2 溶液初始pH的影响
实验条件为:商用Fe3O4用量6.7 g/L(即0.2 g)或天然磁铁矿用量16.7 g/L(即0.5 g),Hg(II)初始浓度为50、200、1 000 μg/L,溶液初始pH为2.0、3.0、5.0、7.0、9.0、11.0(误差为±0.02),反应时间48 h。图2表明商用Fe3O4和天然磁铁矿对Hg(II) 的去除均与pH有很大关系。对于商用Fe3O4,在Hg(II) 初始浓度为200、1 000 μg/L时,pH = 2.0 ~ 5.0范围,Hg(II) 去除率随pH的升高而升高,且在pH>5.0时去除率达到稳定高值;在Hg(II) 初始浓度较低(即50 μg/L)的情况下,Hg(II) 去除率仍然是随pH的升高而升高,但在pH = 3.0以后去除率就基本达到稳定。对于天然磁铁矿,Hg(II) 去除率随pH的变化趋势与商用Fe3O4明显相反,在pH = 2.0 ~ 5.0时,随pH的升高而降低,且在pH>5.0时去除率稳定在较低水平;在Hg(II) 初始浓度50 μg/L时,这种趋势不太明显。
利用visual MINTEQ 3.0模拟实验所用去离子水体系不同pH下的Hg(II)种类分布,输入参数见表4,模拟结果如图 2D所示。结果显示,在 pH<2.2或pH>4.0的溶液中,主要的Hg(II) 种类分别为Hg2+和Hg(OH)2。在pH为1.0 ~ 5.0时,有少量的HgOH+存在,且在pH = 3.0时HgOH+占总Hg(II) 的比例最大为20%。因此,商用Fe3O4对Hg(II) 的去除率之所以随pH的升高而升高并达到稳定高值可能是由于:①如前所述,Hg(II) 可以被Fe3O4中的Fe(II) 还原[19,29]。在低pH时,Fe(II)会被部分溶出,由于水相Fe(II) 对Hg(II) 的还原能力较固相 Fe(II) 低,抑制了 Hg(II)的还原过程[19,30]。②在pH = 2.0 ~ 3.0时,Hg(II) 主要以带正电的Hg2+和Hg(OH)+的形式存在,而由于商用 Fe3O4的零电点(point of zero charge,PZC)是pHpzc = 6.6[31],因此在pH介于2.0 ~ 3.0时商用Fe3O4表面也带正电荷,导致在这个pH范围内的吸附也较低。

图2 不同Hg(II) 初始浓度下pH对Hg(II) 去除率的影响Fig. 2 Effects of initial pH on removal efficiency of Hg (II) with initial concentration of Hg(II)

表4 Visual MINTEQ 3.0模型输入参数Table 4 Input parameters of Visual MINTEQ 3.0
天然磁铁矿的等电点(isoelectric point,IEP)与pHpzc = 5.7[31]差别很大。较高的硅含量导致其pHIEP较低,约为pH = 2.0 ~ 4.2[23–32]。因此在实验pH范围内,天然磁铁矿表面以带负电荷为主,对带正电的Hg2+和Hg(OH)+具有较强的吸引力。随着pH升高,Hg(II) 逐渐变为以中性的 Hg(OH)2为主,因此天然磁铁矿对Hg(II) 的去除率随 pH的升高而降低,在 Hg(OH)2含量达到稳定高值的pH范围内,Hg(II) 去除率也稳定在较低水平。此外,不同 Hg(II) 存在形式与天然磁铁矿表面基团的络合反应也可能导致了这一趋势的产生,但与何种基团发生了何种反应暂不得而知。为避免大量其他离子的引入,在探究其他因素的影响实验中,均不调节溶液的初始pH,即pH= 2.8 ± 0.1。
2.3 吸附剂用量的影响
实验条件为:Hg(II) 初始浓度为200 μg/L,溶液初始pH = 2.8±0.1,反应时间48 h,商用Fe3O4或天然磁铁矿用量为1.67 ~ 166.7 g/L(即0.05 ~ 5.0 g)。图3表明随着吸附剂用量的增加,Hg(II) 去除率相应地增加。商用Fe3O4的用量为33.3 g/L时,Hg(II) 去除率即接近100%,而天然磁铁矿在用量为166.7 g/L时的去除率仅为78%。由于用量>33.3 g/L时,去除率增加缓慢,因此探究其他影响因素时采用33.3 g/L的用量。
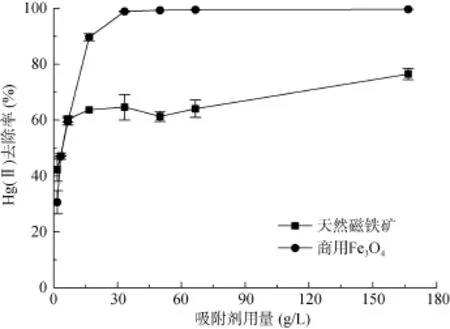
图3 吸附剂用量对Hg(II) 去除率的影响Fig. 3 Effects of sorbent dosages on removal efficiency of Hg(II)
2.4 反应时间的影响
实验条件为:Hg(II) 初始浓度为200 μg/L,商用Fe3O4和天然磁铁矿用量均为 33.3 g/L,溶液初始pH = 2.8±0.1,反应时间5 min ~ 48 h。Hg(II) 去除率随反应时间的变化如图4,在反应初始阶段去除率快速升高,后缓慢上升最后达到平衡,12 h内商用Fe3O4和天然磁铁矿对 Hg(II) 的去除率分别达 97% 和75%。因此后续实验的反应时间设为12 h。
2.5 Hg(II)初始浓度的影响
实验条件为:Hg(II) 初始浓度为50 ~ 2 000 μg/L,商用Fe3O4和天然磁铁矿用量均33.3 g/L,溶液初始pH = 2.8±0.1,反应时间12 h,室温维持在 (27±1)℃。图5表明商用Fe3O4和天然磁铁矿对Hg(II) 的去除率随初始浓度增加而降低,去除量(qe)随初始浓度增加而增加,且 Hg(II) 去除量均是先随初始浓度增加较为快速地上升,而后上升速度减缓。
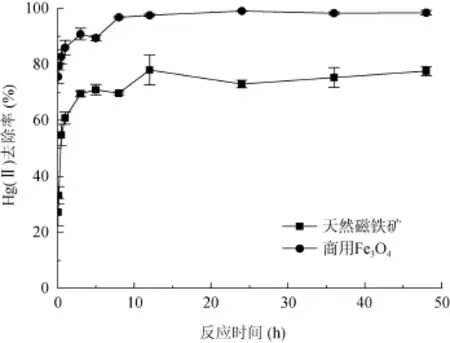
图4 反应时间对Hg(II)去除率的影响Fig. 4 Effects of contact time on removal efficiency of Hg(II)
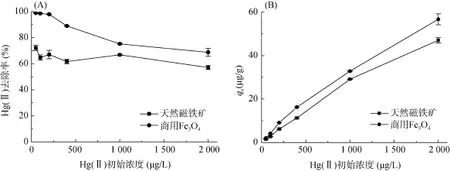
图5 Hg(II)初始浓度对Hg(II) 去除率(A)和去除量(B)的影响Fig. 5 Effects of initial Hg(II) concentration on removal efficiency (A) and removal capacity (B) of Hg (II)
2.6 共存离子的影响
Cl–、SO42–、K+和Ca2+是地下水中常见的阴、阳离子[33–34],因此选择这4种离子来探究其对Hg(II)去除的影响。实验条件为:Hg(II) 初始浓度为200 μg/L,商用Fe3O4和天然磁铁矿用量均为33.3 g/L,溶液初始pH = 2.8±0.1,反应时间12 h,不同浓度的NaCl、Na2SO4、KNO3和Ca(NO3)2·4H2O加入到溶液中用于提供4种离子。
图6表明商用Fe3O4和天然磁铁矿对Hg(II) 的去除受到Cl–的显著影响,在Cl–浓度为50 mg/L时,商用Fe3O4对Hg(II) 的去除率较不含Cl–的体系明显降低,并且随着Cl–浓度增加去除率持续降低,这是由于 Cl–可以与 Hg(II) 形成稳定的水相络合物(如HgCl2、HgCl3–),并成为Hg(II) 的主要存在形式,从而抑制了商用 Fe3O4对 Hg(II) 的吸附过程[35–37]。相反地,天然磁铁矿对 Hg(II) 的去除率在 Cl–浓度为50 mg/L时较Cl–浓度为0 mg/L时升高了20%,这可能是由于形成了金属–配体三元表面络合物≡FeOHgCl[35,38]。在实验的SO42–浓度下,Hg–硫酸盐水相络合物所占比例很低,且不太可能与天然磁铁矿表面形成三元表面络合物[35]。K+和 Ca2+对 Hg(II) 的去除也无显著影响。
2.7 Hg(II)去除机制分析
一些研究表明Hg(II) 在Fe(氢)氧化物上的吸附是由于Hg(II) 与Fe(氢)氧化物表面羟基的络合作用[38–39]。也有报道指出 Cr(VI) 的去除机制是 Cr(VI) 吸附在合成磁铁矿的表面而后又被还原为Cr(III)[40]。本研究将对 Hg(II) 去除的动力学和等温线进行分析,并结合光谱学手段,探究商用 Fe3O4和天然磁铁矿去除Hg(II) 的主要机制。
2.7.1 去除动力学 对不同反应时间的 Hg(II)去除量(qt)用准二级动力学方程[41]进行拟合发现,商用Fe3O4和天然磁铁矿对 Hg(II) 的去除均能很好地符合准二级动力学方程(图 7A)。由准二级动力学方程计算得到的商用 Fe3O4和天然磁铁矿对 Hg(II) 的平衡吸附量分别为5.69 μg/g和4.43 μg/g,十分接近于实验数据(分别为5.67 μg/g和4.47 μg/g),拟合优度R2均大于0.999,说明准二级动力学方程与实验数据具有非常好的相关性。因此,化学吸附是去除Hg(II)的主要速率控制步骤[16,42]。

图6 共存离子Cl–(A)、SO42–(B)、K+(C) 和Ca2+(D) 对Hg(II) 去除率的影响Fig. 6 Effects of coexisting ions of Cl–(A)、SO42–(B)、K+(C) and Ca2+(D) on removal efficiency of Hg(II)

图7 Hg(II)去除的准二级动力学(A)和Freundlich等温模型(B)的线性拟合Fig. 7 Linear fitting of pseudo-second-order kinetic equation (A) and Freundlich isotherm equation (B) of Hg(II) removal
2.7.2 等温线研究 Freundlich等温线的线性表达式如式1所示[10],不同Hg(II) 初始浓度下商用Fe3O4和天然磁铁矿对 Hg(II) 的平衡吸附浓度与平衡吸附量实验数据能够用式(1)很好地拟合(图7B)。

式中:qe(μg/g)和 Ce(μg/L)分别为平衡吸附量和平衡浓度;Kf和n是Freundlich常数,通常分别用来反映吸附量和吸附强度[43]。根据图7B可以看出,对于商用Fe3O4,Kf和1/n分别为2.93和0.43,而对于天然磁铁矿,分别为0.10和0.88。1/n的值均在0和1之间,表明商用 Fe3O4和天然磁铁矿表面的位点均具有异质性,而天然磁铁矿表面的吸附位点更加不均匀[13,44–45]。本研究获得的实验数据用 Langmuir等温模型无法很好地拟合,而Langmuir等温模型描述的是单层吸附,因此暗示了商用Fe3O4和天然磁铁矿对Hg(II)的吸附不是严格的化学吸附[46],物理吸附也起了一定的作用。
2.7.3 吸附前后的FTIR分析 由动力学和等温线研究可知,商用 Fe3O4和天然磁铁矿对 Hg(II) 的去除均是化学吸附和物理吸附的共同作用。基于这一前提,通过对Hg(II) 吸附前后的FTIR图进行比较,研究表面基团的变化[47–48]。设置不同的溶液初始pH和Hg(II) 初始浓度以获得更高的汞吸附量。由图8A可以看出,商用Fe3O4仅在573 cm–1处有一个Fe-O键的吸收峰,表明了商用Fe3O4化学成分单纯[10,49–50]。负载了 Hg(II) 的商用 Fe3O4的红外光谱没有明显变化,表明商用Fe3O4对Hg(II)的吸附没有基团的参与。Wiatrowski等[19]和Pasakarnis等[29]指出Hg(II) 可以被含有Fe(II)/Fe(III) 混合价态的磁铁矿还原,并且还原能力受 Fe(II) 含量的影响。商用 Fe3O4中较高的Fe(II) 含量(21.0% ~ 22.5%)表明Hg(II) 通过物理吸附被吸附到商用Fe3O4表面后可能发生了还原反应。
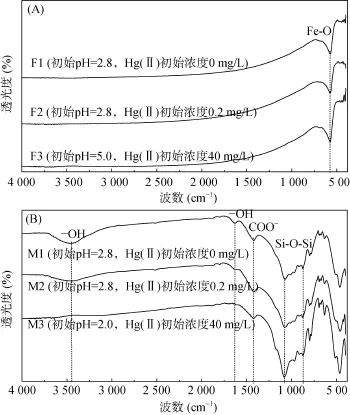
图8 Hg(II) 吸附前后商用Fe3O4(A)与天然磁铁矿(B)的FTIR图谱Fig. 8 FTIR spectra of Fe3O4(A) and natural magnetite (B) prior to and after Hg(II) sorption experiments
由图8B可以看出,天然磁铁矿的吸收峰较多,表明其成分复杂的特性。3 452 cm–1处为 –OH 的伸缩振动[47–48,51],并通常伴随着另一个吸收峰的出现,即1 630 cm–1处吸附水的 –OH振动,1 419 cm–1处为COO−的伸缩振动。由于 Hg(II) 的吸附,天然磁铁矿的表面基团产生了明显变化,吸附后3 452 cm–1处的峰强明显减弱,同时1 630 cm–1处的峰发生了偏移,并且对于高Hg(II) 吸附量的天然磁铁矿样品,3 452 cm–1处的峰消失。这些变化表明羟基是吸附 Hg(II) 的主要官能团[47]。基于Hg(II)在(氢)氧化物上的表面吸附模型(surface complexation model,SCM),推测Hg(II)在天然磁铁矿上的吸附与形成了单配位基的表面络合物 ≡FeOHg+有关,可以用下述公式描述(式2、3、4)[38,52]。因此,天然磁铁矿去除 Hg(II) 的主要机制是 Hg(II) 与表面羟基的络合作用(化学吸附)和物理吸附的共同作用。
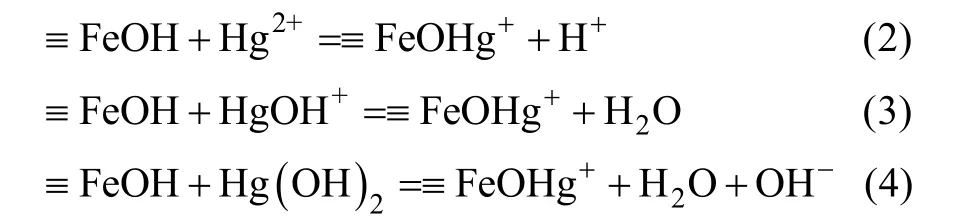
2.7.4 吸附前后的XPS分析 对Hg(II) 吸附前后的天然磁铁矿和商用 Fe3O4样品进行XPS分析,反应条件见表5,其中F0、F1分别代表Hg(II) 吸附前后的商用Fe3O4样品、M0和M1分别代表Hg(II) 吸附前后的天然磁铁矿样品。宽扫描结果(图 9A)表明商用Fe3O4和天然磁铁矿表面都有Fe、O和C的峰,而相对于商用Fe3O4,天然磁铁矿表面的Fe峰较弱,Si峰较强,这些特征也与XRF所得到的元素成分含量相一致。
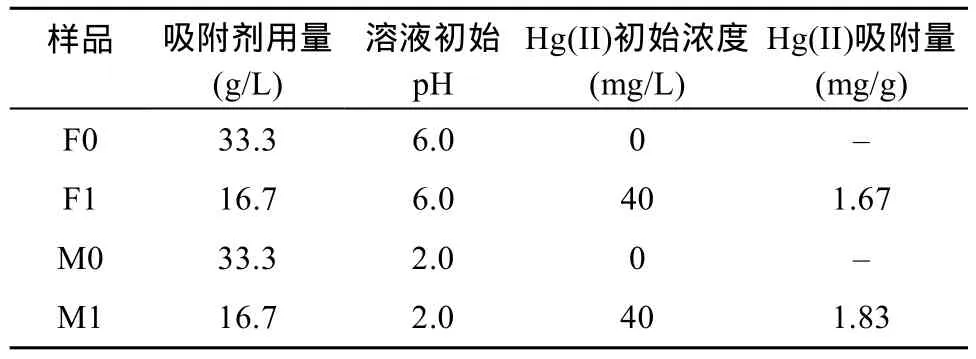
表5 XPS分析的实验条件Table 5 Experimental conditions for XPS analysis
为进一步探究 Hg(II) 的去除机制,对不同实验条件下(表 5)获得的商用 Fe3O4和天然磁铁矿样品的XPS图进行比较,图9A中表明,吸附后的商用Fe3O4和天然磁铁矿表面均未发现 Hg(II)。F1中没有出现Hg的峰可能是由于Hg(II) 被还原后没有持留在商用Fe3O4表面,导致表面的Hg含量较低,没有被检出。M1中也没有检测到Hg的峰。主要由于Si2p和Hg4f的束缚能(binding energy)十分接近,而天然磁铁矿中Si的含量很高,Si2p的峰完全掩盖了Hg4f的峰。因为通过XPS宽扫描图谱无法直接说明Hg的存在及形态,为探究 Hg(II) 去除机制,本研究对吸附前后的Fe2p的窄扫描图(图 9B)进行了分析,目的是通过吸附前后Fe的化学态变化来判断Hg(II) 的吸附方式。
图9B中M0和M1表面Fe2p3/2的峰位于710.4 eV,对应的 Fe的化学态为 Fe2O3或 Fe3O4[53–54],吸附了Hg之后峰位没有明显变化,说明Fe的化学态变化可以忽略。F0表面Fe2p3/2的峰位于710.7 eV,对应Fe2O3或Fe3O4[55–57],然而吸附了Hg之后,F1表面Fe2p3/2的峰偏移到709.9 eV,对应的是Fe2O3[58],一定程度上可以反映出Fe(II) 参与了Hg(II) 的还原并形成了Fe(III)。但是,本研究发现,由于较低的Hg(II) 吸附量、天然磁铁矿中较高的杂质含量,很难通过常规的光谱分析仪器定量化学和物理吸附所占的比例,今后的研究中若能够借助同步辐射技术将对 Hg(II) 去除机制的认识有很大帮助。

图9 Hg(II) 吸附前后商用Fe3O4与天然磁铁矿的XPS宽扫描图谱 (A) 和Fe2p的XPS窄扫描图 (B)Fig. 9 XPS spectra (A) and Fe2p XPS spectra of magnetites (B) prior to and after sorption experiments
2.8 模拟地下水中Hg(II) 的吸附实验
通过不同吸附剂用量对模拟地下水体系中Hg(II)去除率的影响,探究了天然磁铁矿和商用 Fe3O4在Hg(II) 污染模拟地下水中的有效性。实验条件为:Hg(II) 初始浓度为200 μg/L,溶液体积30 ml,模拟地下水溶液,初始pH = 7.0,反应时间48 h,商用Fe3O4和天然磁铁矿用量为1.67 ~ 166.7 g/L(即0.05 ~ 5.0 g)。图10显示商用Fe3O4和天然磁铁矿对模拟地下水中Hg(II) 的最大去除率分别达95% 和90%,表明二者均具有修复Hg(II) 污染地下水的潜力。
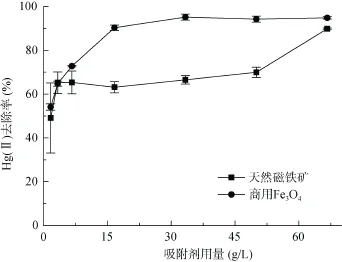
图10 吸附剂用量对模拟地下水中Hg(II)去除率的影响Fig. 10 Effects of sorbent dosages on removal efficiency of Hg(II) from simulated groundwater
2.9 模拟地下水中Hg(II) 的脱附实验
利用模拟地下水对吸附 Hg(II)的天然磁铁矿和商用Fe3O4进行了脱附研究。脱附率(D,%)可由下式计算得到:

式中:C0和Ce分别为溶液中Hg(II) 的初始浓度和平衡浓度(μg/L),Ce(D)为脱附溶液中 Hg(II) 的平衡浓度(μg/L)。脱附实验之前需对天然磁铁矿和商用Fe3O4进行吸附Hg(II) 的处理,吸附实验条件为:模拟地下水体系,Hg(II) 初始浓度为200 μg/L,pH = 7.0±0.1,商用Fe3O4和天然磁铁矿用量均为33.3 g/L。反应72 h后,商用Fe3O4和天然磁铁矿对Hg(II) 的去除率分别为 76.6% 和 76.2%。将吸附后的固体过滤分离,在模拟地下水中脱附60 h后,脱附溶液中Hg(II) 的残余浓度分别为12.6 μg/L和2.2 μg/L。利用公式 5计算可知吸附Hg(II) 的商用Fe3O4和天然磁铁矿的Hg(II) 脱附率分别为8.3% 和1.4%,表明二者在地下水中均能较为稳定地存在,并且天然磁铁矿较商用Fe3O4更为稳定。
3 结论
1) 商用 Fe3O4和天然磁铁矿均能够有效地去除Hg(II),去除率受pH、用量、反应时间、Hg(II)初始浓度、Cl–等因素的影响,但影响程度和趋势不尽相同:随用量的增加、反应时间的延长、Hg(II)初始浓度的升高,二者的Hg(II) 去除率均上升;但随pH的升高,商用 Fe3O4对 Hg(II) 的去除率升高,天然磁铁矿对 Hg(II) 的去除率降低;Cl–的存在使得商用Fe3O4对Hg(II) 的去除率明显降低,而天然磁铁矿对Hg(II) 的去除率有所上升。
2) 商用Fe3O4和天然磁铁矿对Hg(II) 的去除均能很好地均符合准二级动力学模型和 Freundlich模型,说明化学吸附是主要的速率控制步骤,同时也有物理吸附的作用。结合光谱分析,可知商用Fe3O4对Hg(II) 的去除机制主要是化学还原和物理吸附;天然磁铁矿去除 Hg(II) 的机制主要是羟基络合与物理吸附。
3) 商用 Fe3O4和天然磁铁矿对模拟地下水中Hg(II) 的去除率分别达95% 和90%,二者均具有地下水修复的应用潜力。且天然磁铁矿的 Hg(II) 脱附率较商用Fe3O4低,在地下水中能够更为稳定地存在。本研究表明天然磁铁矿稳定性更好、具有十分明显的成本优势以及在相同用量的条件下与商用 Fe3O4的Hg(II) 去除率差别不大,因此天然磁铁矿适合用作PRB活性填充材料。
[1] 胡月红. 国内外汞污染分布状况研究综述[J]. 环境保护科学, 2008(1): 38–41
[2] Gray J E, Theodorakos P M, Bailey E A, et al. Distribution, speciation, and transport of mercury in stream-sediment, stream-water, and fish collected near abandoned mercury mines in southwestern Alaska, USA[J]. Science of the Total Environment, 2000, 260(1): 21–33
[3] Wang J X, Feng X B, Anderson C W N, et al. Remediation of mercury contaminated sites—A review[J]. Journal of Hazardous Materials, 2012, 221: 1–18
[4] 冯新斌, 仇广乐, 付学吾, 等. 环境汞污染[J]. 化学进展, 2009(Z1): 436–457
[5] Blue L Y, Van Aelstyn M A, Matlock M, et al. Low-level mercury removal from groundwater using a synthetic chelating ligand[J]. Water Research, 2008, 42(8–9): 2025–2028
[6] 薛之化. 电石法聚氯乙烯生产中的汞污染治理[J]. 中国氯碱, 2011(2): 25–31
[7] Bollen A, Wenke A, Biester H. Mercury speciation analyses in HgCl2-contaminated soils and groundwater – Implications for risk assessment and remediation strategies[J]. Water Research, 2008, 42(1–2): 91–100
[8] Campbell K R, Ford C J, Levine D A. Mercury distribution in poplar creek, Oak Ridge, Tennessee, USA[J]. Environmental Toxicology and Chemistry, 1998, 17(7): 1191–1198
[9] Qiu G L, Feng X B, Meng B, et al. Environmental geochemistry of an abandoned mercury mine in Yanwuping, Guizhou Province, China[J]. Environmental Research, 2013, 125: 124–130
[10] Zhang S X, Zhang Y Y, Liu J S, et al. Thiol modified Fe3O4@SiO2as a robust, high effective, and recycling magnetic sorbent for mercury removal[J]. Chemical Engineering Journal, 2013, 226: 30–38
[11] Goel J, Kadirvelu K, Rajagopal C. Mercury (II) removal from water by coconut shell based activated carbon: Batch and column studies[J]. Environmental Technology, 2004, 25(2): 141–153
[12] Yardim M F, Budinova T, Ekinci E, et al. Removal of mercury (II) from aqueous solution by activated carbon obtained from furfural[J]. Chemosphere, 2003, 52(5): 835–841
[13] Zhang F S, Nriagu J O, Itoh H. Mercury removal from water using activated carbons derived from organic sewage sludge[J]. Water Research, 2005, 39(2–3): 389–395
[14] Arsuaga J M, Aguado J, Arencibia A, et al. Aqueous mercury adsorption in a fixed bed column of thiol functionalized mesoporous silica[J]. Adsorption-Journal of the International Adsorption Society, 2014, 20(2–3): 311–319
[15] Liu J F, Zhao Z S, Jiang G B. Coating Fe3O4magnetic nanoparticles with humic acid for high efficient removal of heavy metals in water[J]. Environmental Science & Technology, 2008, 42(18): 6949–6954
[16] Pan S D, Shen H Y, Xu Q H, et al. Surface mercapto engineered magnetic Fe3O4nanoadsorbent for the removal of mercury from aqueous solutions[J]. Journal of Colloid and Interface Science, 2012, 365(1): 204–212
[17] 潘胜东. 巯基功能化纳米 Fe3O4磁性高分子复合材料的合成、表征及其对水中 Hg(II)的吸附研究[D]. 杭州: 浙江大学, 2012
[18] 宋娟, 李晓燕, 张升晓, 等. 磁性 Fe3O4@SiO2-SH 的制备及环境水体中汞的富集与分析[J]. 分析试验室, 2014(12): 1374–1377
[19] Wiatrowski H A, Das S, Kukkadapu R, et al. Reduction of Hg(II) to Hg(0) by magnetite[J]. Environmental Science & Technology, 2009, 43(14): 5307–5313
[20] 赵谨. 天然磁铁矿与褐铁矿处理含Hg(II)、Cd(II)、Cr(II)废水实验研究[D]. 北京: 中国地质大学(北京), 2003
[21] 成翠兰, 毋伟, 沈淑玲, 等. 纳米四氧化三铁吸附水中汞离子的研究[J]. 北京化工大学学报(自然科学版), 2008 (3): 5–8
[22] Lamborg C H, Kent D B, Swarr G J, et al. Mercury speciation and mobilization in a wastewater-contaminated groundwater plume[J]. Environmental Science & Technology, 2013, 47(23): 13239–13249
[23] Salazar-Camacho C, Villalobos M, Rivas-Sanchez M D, et al. Characterization and surface reactivity of natural and synthetic magnetites[J]. Chemical Geology, 2013, 347: 233–245
[24] Liu Y, Mou H, Chen L, et al. Cr(VI)-contaminated groundwater remediation with simulated permeable reactive barrier (PRB) filled with natural pyrite as reactive material: Environmental factors and effectiveness[J]. Journal of Hazardous Materials, 2015, 298: 83–90
[25] Oliva J, De Pablo J, Cortina J L, et al. Removal of cadmium, copper, nickel, cobalt and mercury from water byApatite II™: Column experiments[J]. Journal of hazardous materials, 2011, 194: 312–323
[26] Weisener C G, Sale K S, Smyth D J A, et al. Field column study using zerovalent iron for mercury removal from contaminated groundwater[J]. Environmental Science & Technology, 2005, 39(16): 6306–6312
[27] Murphy E M, Ginn T R, Chilakapati A, et al. The influence of physical heterogeneity on microbial degradation and distribution in porous media[J]. Water Resources Research, 1997, 33(5): 1087–1103
[28] 中华人民共和国环境保护部. 水质 汞、砷、硒、铋和锑的测定原子荧光法[S]. 北京: 中国环境科学出版社, 2014
[29] Pasakarnis T S, Boyanov M I, Kemner K M, et al. Influence of chloride and Fe(II) content on the reduction of Hg(II) by magnetite[J]. Environmental Science & Technology, 2013, 47(13): 6987–6994
[30] Charlet L, Bosbach D, Peretyashko T. Natural attenuation of TCE, As, Hg linked to the heterogeneous oxidation of Fe(II): An AFM study[J]. Chemical Geology, 2002, 190(1–4): 303–319
[31] Kosmulski M. Compilation of PZC and IEP of sparingly soluble metal oxides and hydroxides from literature[J]. Advances in Colloid and Interface Science, 2009, 152(1–2): 14–25
[32] Somasundaran P, AIME. Fine particles processing: proceedings of the international symposium on fine particles processing, Las Vegas, Nevada, February 24–28, 1980[M]. Englewood, CO, USA: American Institute of Mining, Metallurgical, and Petroleum Engineers, 1980
[33] 杨文君, 郭迎庆, 杜尔登. 地下水中常见离子对纳米零价铁除 Se(IV)动力学的影响[J]. 环境科学, 2014(5): 1793–1797
[34] 张燕, 刘宏远, 陈英旭. 地下水中常见离子对催化还原去除硝酸盐的影响[J]. 农业环境科学学报, 2006(4): 1071–1075
[35] Kim C S, Rytuba J, Brown G E. EXAFS study of mercury(II) sorption to Fe- and Al-(hydr)oxides - II. Effects of chloride and sulfate[J]. Journal of Colloid and Interface Science, 2004, 270(1): 9–20
[36] Barrow N J, Cox V C. The effects of pH and chloride concentration on mercury sorption. I. by goethite[J]. Journal of Soil Science, 1992, 43(2): 295–304
[37] Morse J W, Luther G W. Chemical influences on trace metal-sulfide interactions in anoxic sediments[J]. Geochimica Et Cosmochimica Acta, 1999, 63(19–20): 3373–3378
[38] Bonnissel-Gissinger P, Alnot M, Lickes J P, et al. Modeling the adsorption of mercury(II) on (hydr)oxides II: alpha-FeOOH (goethite) and amorphous silica[J]. Journal of Colloid and Interface Science, 1999, 215(2): 313–322
[39] Kinniburgh D, Jackson M. Adsorption of mercury (II) by iron hydrous oxide gel[J]. Soil Science Society of America Journal, 1978, 42(1): 45–47
[40] Jung Y, Choi J, Lee W. Spectroscopic investigation of magnetite surface for the reduction of hexavalent chromium[J]. Chemosphere, 2007, 68(10): 1968–1975
[41] Ho Y S, McKay G. Pseudo-second order model for sorption processes[J]. Process Biochemistry, 1999, 34(5): 451–465
[42] Ho Y S. Review of second-order models for adsorption systems[J]. Journal of Hazardous Materials, 2006, 136(3): 681–689
[43] Xiong C, Li Y, Wang G, et al. Selective removal of Hg (II) with polyacrylonitrile-2-amino-1, 3, 4-thiadiazole chelating resin: Batch and column study[J]. Chemical Engineering Journal, 2015, 259: 257–265
[44] Etale A, Yalala B, Tutu H, et al. Adsorptive removal of mercury from acid mine drainage: a comparison of silica and maghemite nanoparticles[J]. Toxicological and Environmental Chemistry, 2014, 96(4): 542–554
[45] Mishra S P, Tiwari D, Dubey R S, et al. Biosorptive behaviour of casein for Zn2+, Hg2+and Cr3+: Effects of physico-chemical treatments[J]. Bioresource Technology, 1998, 63(1): 1–5
[46] Bao J, Fu Y, Bao Z H. Thiol-functionalized magnetite/ graphene oxide hybrid as a reusable adsorbent for Hg2+removal[J]. Nanoscale Research Letters, 2013, 8 (1): 1
[47] Dong X L, Ma L N Q, Zhu Y J, et al. Mechanistic investigation of mercury sorption by Brazilian pepper biochars of different pyrolytic temperatures based on X-ray photoelectron spectroscopy and flow calorimetry[J]. Environmental Science & Technology, 2013, 47(21): 12156–12164
[48] Elangovan R, Philip L, Chandraraj K. Biosorption of hexavalent and trivalent chromium by palm flower (Borassus aethiopum)[J]. Chemical Engineering Journal, 2008, 141(1–3): 99–111
[49] Singh S, Barick K C, Bahadur D. Surface engineered magnetic nanoparticles for removal of toxic metal ions and bacterial pathogens[J]. Journal of Hazardous Materials, 2011, 192(3): 1539–1547
[50] Sun Z X, Su F W, Forsling W, et al. Surface characteristics of magnetite in aqueous suspension[J]. Journal of Colloid and Interface Science, 1998, 197(1): 151–159
[51] Johari K, Saman N, Mat H. A comparative evaluation of mercury(II) adsorption equilibrium and kinetics onto silica gel and sulfur-functionalised silica gels adsorbents[J]. Canadian Journal of Chemical Engineering, 2014, 92(6): 1048–1058
[52] Sarkar D, Essington M E, Misra K C. Adsorption of mercury(II) by variable charge surfaces of quartz and gibbsite[J]. Soil Science Society of America Journal, 1999, 63(6): 1626–1636
[53] Nefedov V I, Salyn Y V, Leonhardt G, et al. Comparison of different spectrometers and charge corrections used in X-ray photoelectron-spectroscopy[J]. Journal of Electron Spectroscopy and Related Phenomena, 1977, 10(2): 121–124
[54] Oku M, Hirokawa K. X-ray photoelectron-spectroscopy ofCo3O4, Fe3O4, Mn3O4and related compounds[J]. Journal of Electron Spectroscopy and Related Phenomena, 1976, 8(6): 475–481
[55] Hawn D D, Dekoven B M. Deconvolution as a correction for photoelectron inelastic energy-losses in the core level XPS spectra of iron-oxides[J]. Surface and Interface Analysis, 1987, 10(2–3): 63–74
[56] Tan B J, Klabunde K J, Sherwood P M A. X-ray photoelectron-spectroscopy studies of solvated metal atom dispersed catalysts-monometallic iron and bimetallic iron cobalt particles on alumina[J]. Chemistry of Materials, 1990, 2(2): 186–191
[57] Brion D. Study by photoelectron-spectroscopy of surface degradation of FeS2, CuFeS2, ZnS and PbS exposed to air and water[J]. Applications of Surface Science, 1980, 5(2): 133–152
[58] Paparazzo E. XPS and auger spectroscopy studies on mixtures of the oxides SiO2, Al2O3, Fe2O3, and Cr2O3[J]. Journal of Electron Spectroscopy and Related Phenomena, 1987, 43(2): 97–112
Simulating Remediation of Hg(II)-Contaminated Groundwater Using Natural Magnetite and Commercial Fe3O4
LIN Na1,2, SONG Xin1*, GUO Liang3, YIN Penghua4, HU Zhihao4
(1 Key Laboratory of Soil Environment and Pollution Remediation, Institute of Soil Science, Chinese Academy of Sciences, Nanjing 210008, China; 2 University of Chinese Academy of Sciences, Beijing 100049, China; 3 Nanjing Kangdi Environmental Protection Technology Co., Ltd., Nanjing 210002, China; 4 College of Engineering and Applied Science, Nanjing University, Nanjing 210093, China)
Currently, remediation of Hg(II)-contaminated groundwater is technically challenging and very costly. It is urgently needed to develop effective and cost-effective technology and material to remediate Hg(II)-contaminated groundwater. The efficiency and mechanism of Hg(II) removal from groundwater by two types of iron–based sorbents, natural magnetite and commercial Fe3O4, were investigated in this study. Hg(II) sorption and desorption on magnetites were also investigated in simulated groundwater. The results showed that both natural magnetite and commercial Fe3O4removed Hg(II) effectively, and Hg(II) removal efficiency was influenced by initial pH, initial Hg(II) concentration, coexisting ion of Cl–, and so on. Sorption kinetic data were well fitted to the pseudo-second-order model. The sorption isotherm could be described by the Freundlich model. The sorption of Hg(II) on natural magnetite was mainly due to the complexation between Hg(II) and surface hydroxyl groups and physisorption, while the sorption of Hg(II) on commercial Fe3O4was largely due to the reduction of Hg(II) by Fe(II) in Fe3O4and physisorption. Hg(II) removal from simulated groundwater by natural magnetite and commercial Fe3O4were nearly 90% and 95%, respectively, demonstrating the application potential of both sorbents in remediating Hg(II)-contaminated groundwater.
Natural magnetite; Commercial Fe3O4; Remediation; Hg(II); Groundwater contamination
X53
A
10.13758/j.cnki.tr.2017.01.018
中国科学院重点部署项目(KZZD-EW-TZ-13)资助。
* 通讯作者(xsong@issas.ac.cn)
林娜(1992—),女,安徽亳州人,硕士研究生,主要从事土壤和地下水污染修复技术与原理研究。E-mail: nlin@issas.ac.cn
猜你喜欢
中国资源综合利用(2022年9期)2022-10-13
汽车实用技术(2022年13期)2022-07-19
——徐小林
孙子研究(2022年2期)2022-06-09
现代矿业(2022年3期)2022-04-09
新疆钢铁(2021年1期)2021-10-14
商用汽车(2021年12期)2021-07-14
有色设备(2021年4期)2021-03-16
制造技术与机床(2019年9期)2019-09-10
中南大学学报(自然科学版)(2016年2期)2017-01-19
IT时代周刊(2015年7期)2015-11-11
