
Diverse coagulopathies in a rabbit model with different abdominal injuries
2017-04-20 01:42:49RuoWuLuogenPengHuiminZhao
Ruo Wu, Luo-gen Peng, Hui-min Zhao
1Department of Emergency Medicine, Haikou People’s Hospital Aff i liated to Central South University, Haikou 570208, China
2Department of Emergency Medicine, the First Aff i liated Hospital of Guangxi Medical University, Nanning 530021, China
Diverse coagulopathies in a rabbit model with different abdominal injuries
Ruo Wu1, Luo-gen Peng2, Hui-min Zhao2
1Department of Emergency Medicine, Haikou People’s Hospital Aff i liated to Central South University, Haikou 570208, China
2Department of Emergency Medicine, the First Aff i liated Hospital of Guangxi Medical University, Nanning 530021, China
BACKGROUND: Although coagulopathy can be very common in severe traumatic shock patients, the exact incidence and mechanism remain unclear. In this study, a traumatic shock rabbit model with special abdomen injuries was developed and evaluated by examining indicators of clotting and fi brinolysis.
METHODS: Forty New Zealand white rabbits were randomly divided into four groups: group 1 (sham), group 2 (hemorrhage), group 3 (hemorrhage-liver injury), and group 4 (hemorrhage-liver injury/intestinal injury-peritonitis). Coagulation was detected by thromboelastography before trauma (T0), at 1 hour (T1) and 4 hours (T2) after trauma.
RESULTS: Rabbits that suffered from hemorrhage alone did not differ in coagulation capacity compared with the sham group. The clot initiations (R times) of group 3 at T1and T2were both shorter than those of groups 1, 2, and 4 (P<0.05). In group 4, clot strength was decreased at T1and T2compared with those in groups 1, 2, and 3 (P<0.05), whereas the R time and clot polymerization were increased at T2(P<0.05). The clotting angle signif i cantly decreased in group 4 compared with groups 2 and 3 at T2(P<0.05).
CONCLUSION: This study suggests that different abdominal traumatic shock show diverse coagulopathy in the early phase. Isolated hemorrhagic shock shows no obvious effect on coagulation. In contrast, blunt hepatic injury with hemorrhage shows hypercoagulability, whereas blunt hepatic injury with hemorrhage coupled with peritonitis caused by a ruptured intestine shows a tendency toward hypocoagulability.
Hemorrhagic shock; Multiple trauma model; Inflammation; Coagulopathy; Thromboelastography
INTRODUCTION
Trauma is the leading cause of death worldwide; hemorrhage is a major contributor to this mortality and accounts for almost 50% of the deaths within the initial 24 hours.[1]Approximately 24% to 34% of trauma patients present with coagulopathy when they arrive in the emergency department.[2,3]Although traumatic coagulopathy may appear in an early or delayed form, the patients with traumatic shock have signif i cantly different morbidity of coagulopathy.[4,5]Trauma-associated coagulopathy was originally thought to be primarily due to hypothermia, acidosis and consumption/dilution of clotting factors.[6]Current studies have revealed that neurohormonal activation, systemic inflammation and widespread endothelial damage may similarly have an important influential role, and the etiology of traumarelated coagulopathy is multifactorial.[7,8]Much mystery still surrounds changes in coagulation taking place in the period after injury. This study developed a rabbit model of traumatic shock with abdominal injuries to characterizethe changes in clotting and fi brinolysis function by whole blood thromboelastography. We hypothesize that changes in hemostatic function will be present shortly after trauma and will change as trauma severity increases.
METHODS
Animal care
This study was approved by the Institutional Ethics Committee of Guangxi Medical University. Forty New Zealand rabbits (male and female unlimited), aged 8–10 weeks, weighing between 2.30–2.65 kg, were provided by the Experimental Animal Center of Guangxi Medical University (registration number SCXK 2009–0002). The rabbits were equally randomized (simple randomization) into one of the following four groups: group 1 (sham), group 2 (hemorrhage), group 3 (hemorrhage-liver injury), or group 4 (hemorrhage-liver injury/intestinal injury-peritonitis). The blood samples were collected before trauma (T0), at 1 hour (T1) and 4 hours (T2) after trauma. The rabbits had unlimited access to food and water before the experiment.
Animal preparation
Animals were intravenously anesthetized with 20% urethane (1 g/kg, Duly, Nanjing, China) via ear vein, and additional doses (0.2 g/kg) were administered as needed. The animals were maintained in a supine position. A temperature probe (MT-JC218, Shinland, Shenzhen, China) was placed rectally for continuous measurement of central body temperature. The left carotid artery was cannulated to allow for continuous recording of the mean arterial pressure (BL-420F, Bio-Medical Electronics Co., Chendu, China), blood laboratory sampling, and bleeding induction. The right internal jugular vein was cannulated for fluid resuscitation. Sham animals were only anesthetized and subjected to arterial, venous, and sham laparotomies.
Multiple injuries and resuscitation
In group 3, a sterile laparotomy was performed via the right lower costal margin, and the liver was gently exposed. The right upper liver blunt-impact injury was established by hitting the exposed liver with a self-made iron bar (diameter=22 mm, length=39.5 mm, weight=100 g) from a height of 20 centimeters, according to AAST scale,[9]the liver injury grade was grade II. Group 4 also underwent blunt hepatic trauma, and the rabbits were subjected to five intestinal crushes and the injection of 5 mL of self-liquid feces into the peritoneal cavity to induce chemical peritonitis. The wound was temporarily closed with sutures. Finally, pressure-controlled hemorrhage was induced in groups 3 and 4 by bleeding approximately 33% of the total blood volume over a 15-minute period until a mean arterial blood pressure (ABP) of 40 mmHg was reached. ABP of 40 mmHg was maintained by controlling the carotid artery bloodletting. Blood pressure was allowed to fl uctuate between 35 and 45 mmHg. Group 2 underwent only controlled hemorrhage and laparotomy. Hemorrhagic shock was maintained for 1 hour. The hemorrhage volume in trauma groups was 57.6±8.0 mL.
Fluid resuscitation was administered to the trauma groups (2, 3, and 4) at T1, adjust speed with reference to ABP target 50–60 mmHg.[10]Fluid administration used hydroxyethyl starch 130/0.4 and a sodium chloride injection (6.9%, Kelun, Sichuan, China) at a volume equivalent to the blood loss volume. Fluid resuscitation was also administered to the sham group and was performed with the mean fl uid volume of the trauma groups.
Blood samples
Blood samples were collected from the left carotid artery. Routine blood examination (e.g., platelets, hemoglobin, and NEUT%) was performed from 1 mL EDTA tubes (Jingzi, Jiangxi, China) with the URIT 3010 (Urit, Guilin, China). Samples for alanine aminotransferase (ALT) and lactate measurements were drawn into 4 mL vacuum tubes (Jingzi, Jiangxi, China) and 1 mL EDTA tubes respectively and were analyzed with the Hitachi automated biochemistry analyzer 7600 (Hitachi, Tokyo, Japan). Samples for thromboelastography (TEG 5000, Haemoscope, Niles, Illinois, America) were collected in 2 mL citrate tubes (Jingzi, Jiangxi, China).
Statistical analysis
Statistical analyses were performed using SPSS 16.0 software (SPSS, Inc., USA). The experimental data were expressed as mean±SD. One-way ANOVA was used to compare multiple groups and within a group between different time points. A difference with P<0.05 was considered signif i cant.
RESULTS
Laboratory assessments
At the beginning of the experiment, physiologic variables were not different significantly among the groups (Table 1). At T1, trauma resulted in comparableimpairment of physiologic variables in the three trauma groups (2, 3, and 4) compared with the sham group, with decreases in temperature, MAP and hemoglobin (P<0.05). Additionally, the hemoglobin levels in groups 3 and 4 were significantly decreased compared with group 2 (P<0.05), and the temperature of group 4 was significantly lower than that of group 2 (P<0.05). In all trauma groups (2, 3, and 4), the lactate levels increased from baseline and were signif i cantly higher than those in group 1 (P<0.05).
At T2, temperature, MAP and hemoglobin continuously decreased in the trauma groups (2, 3, and 4) until the end of the observation period, and the temperature of group 4 was significantly decreased compared with the other groups (P<0.05). The lactate values remained increased for the trauma groups (2, 3, and 4) compared with the sham group, and the ALT levels of groups 3 and 4 were significantly higher than those of groups 1 and 2 (P<0.05). The lactate and NEUT% values were increased significantly in group 4 and were significantly different from those of the other groups (P<0.05).
Two rabbits in group 4 died within 3.7 hours; blood samples were collected from the dying rabbits, and the values were incorporated into the T2results.
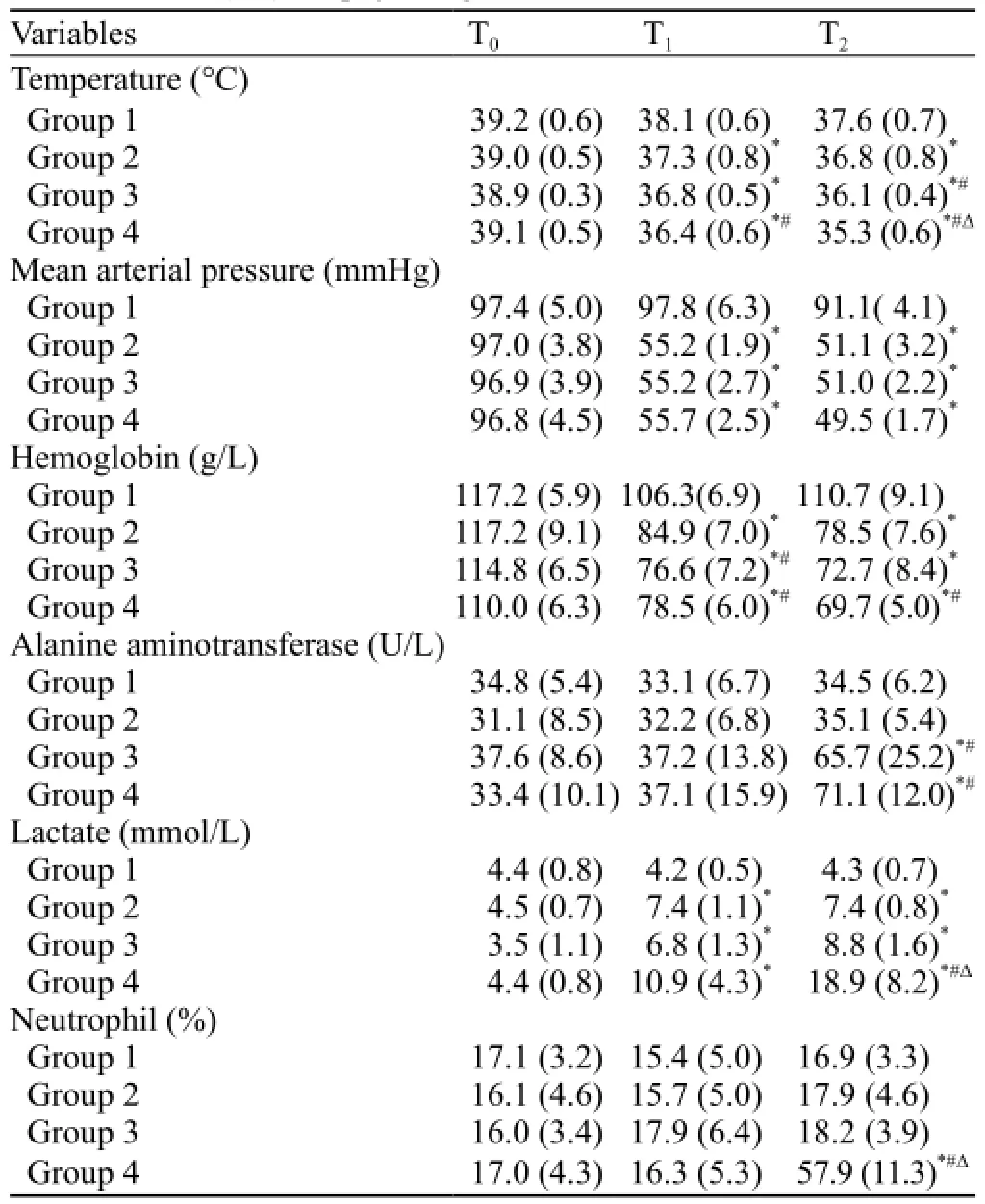
Table 1. Mean (SD) for physiologic variables
Thromboelastography
Before inducing the trauma, the thromboelastography values were not significantly different among the four groups.
Following trauma, clot initiation (R times) in group 3 was signif i cantly shorter than that of the other groups (P<0.05) at T1and continuously shortened until the end of the observation period (P<0.05, T2). The R time was signif i cantly prolonged in group 4 compared with groups 1, 2, and 3 (P<0.05, T2; Figure 1A).
Clot polymerization (K times) in group 4 was signif i cantly longer than that of the other groups (P<0.05, T2). No difference was observed among groups 1, 2, and 3 (Figure 1B).
Clotting angle (Ang) signif i cantly decreased in group 4 compared with groups 2 and 3 (P<0.05, T2; Figure 1C).
Clot strength (MA) signif i cantly decreased in group 4 compared with that of the other groups (P<0.05) at T1and continuously decreased through T2(P<0.05; Figure 1D).
In groups 1 and 2, the thromboelastography values were not significantly different among the three time points. In group 3, the R and K times at T1and T2were significantly shorter than those at T0, and Ang signif i cantly increased at T2compared with T0(P<0.05). In group 4, the R and K times at T2significantly prolonged as compared with those at T0, and MA significantly decreased at T1compared with T0and continuously decreased through T2(P<0.05, Table 2).
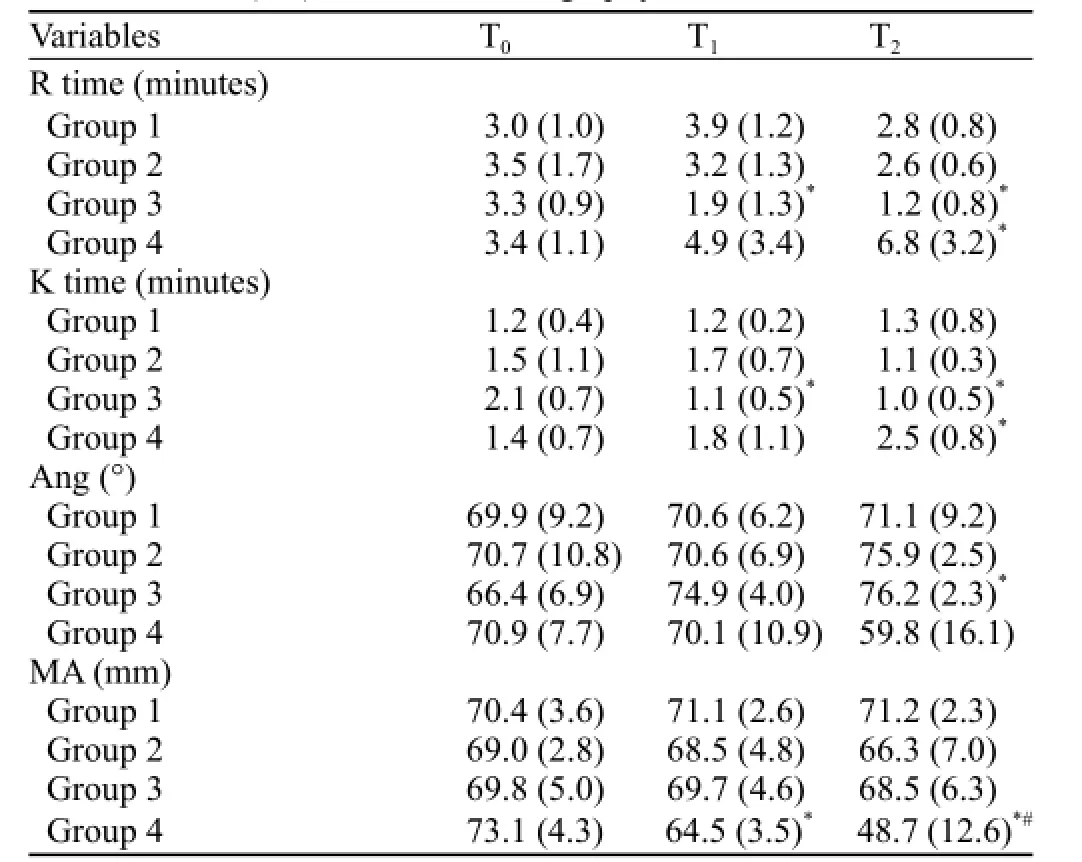
Table 2. Mean (SD) for thromboelastgraphy values
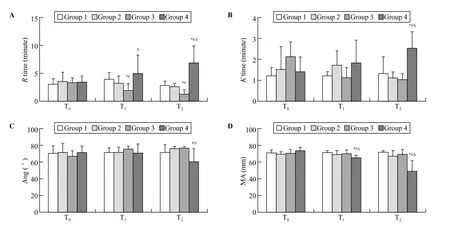
Figure 1. Mean (SD) for thromboelastography values. T0: baseline; T1: after trauma 1 hour; T2: at the end of the experiment; R times: clot initiation; K times: clot polymerization; Ang: clotting angle; MA: clot strength; when compared with group 1,*P<0.05; when compared with group 2,#P<0.05; when compared with group 3,ΔP<0.05.
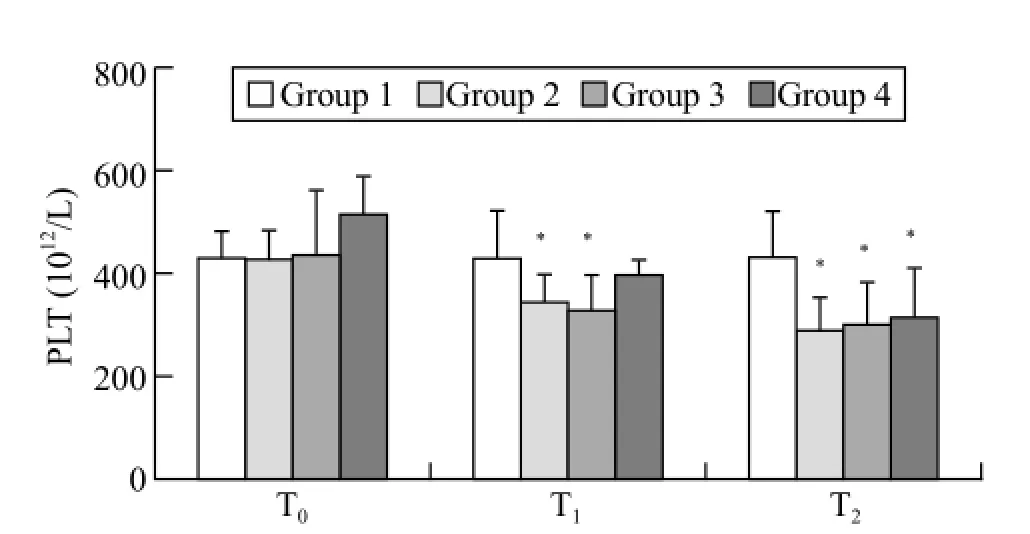
Figure 2. Mean (SD) for platelets. T0: baseline; T1: after trauma 1 hour; T2: at the end of the experiment; when compared with group 1,*P<0.05.
Platelet count
The baseline platelet counts were not different among the groups. Following trauma induction, the platelet counts in trauma groups 2 and 3 were significantly decreased compared with that in the sham group (P<0.05, T1). At the end of the observation period, the platelet counts in all trauma groups (2, 3, and 4) were significantly decreased compared with the sham group (P<0.05, T2; Figure 2).
Macroscopic results
After experiment, careful exploration of the injured liver and intestines were performed. In groups 3 and 4, we were not able to detect macroscopic or petechial bleeding in the peritoneal cavity.
DISCUSSION
Injured patients who develop coagulopathy have an increased mortality risk, but the pathogenesis of traumatic coagulopathy is still not fully elucidated. The primary reasons were originally thought to be the loss of hemostatic factors resulting from severe hemorrhage and consumption, dilution by fluid resuscitation, hypoperfusion and acidosis attributed to shock.[11]The current explanation of traumatic coagulopathy is based on the concept of homeostasis; when hemostatic balances were breakdown, the phenotype of coagulation is depending on the injury, shock, or infection.[12]Our study shows that nontraumatic hemorrhagic shock has no obvious effect on blood coagulation, blunt hepatic injury with hemorrhage induces hypercoagulability, and blunt hepatic injury with hemorrhage coupled with peritonitis caused by a ruptured intestine shows a tendency toward hypocoagulability. Thus, it remains unclear whether traumatic coagulopathy is correlated with injury, infection and inf l ammation.
In this study, the hemorrhage volume in trauma groups was approximately 30% to 35% of the rabbit blood volume[13]and is equivalent to adult severe shock.[14]Hemorrhagic shock was the only trauma factor in group 2; therefore, we could observe the change of coagulation in the pathological state of shock. In our study, group 2 had a remarkably decreased platelet count, lowertemperature and acidosis. However, coagulation was not significantly changed until the end of T2. To date, only a few studies have examined coagulation in isolated experimental hemorrhagic shock models. Fung et al[15]also evaluated nontraumatic hemorrhage without impairing coagulation in an ovine model. Some reports have shown that hemorrhagic shock may play an integral role in the progression of early coagulopathy,[16,17]but our results revealed that hypoperfusion, lower temperature and acidosis may exacerbate the development of coagulopathy. Coagulation dysfunction was not initiated by the isolated hemorrhagic shock.
In groups 3 and 4, the injuries of trauma and shock were combined, but the clotting function changed signif i cantly. During the experiment, the R time was shorter in group 3 than in other groups, whereas temperature, lactate and platelet count were comparable. Mulier et al[18]also found an increased rate of clot formation in a swine multitrauma and hemorrhage study. With minor trauma, tissue-type plasminogen activator (tPA) and activation of the protein C (PC) pathway could counterbalance the hemostatic activation, and the coagulation display a normal phenotype.[19]However, with increasing tissue trauma, the progressively increasing catecholamine level further enhances thrombin generation, which exceeds the released level of tPA and PC; the net hemostatic effect is hypercoagulability.[19,20]This pathway may underlie the changes in coagulation function in group 3. In group 4, both R time and K time were signif i cantly prolonged compared with those of the other groups, and MA significantly decreased. In addition, experimental results similar to ours were found in the study reported by Martini et al.[21,22]Regarding trauma duration, in group 4, the R and K times at T2signif i cantly prolonged as compared with those at T0, and MA continuously decreased through T2. It suggests the impact of intestinal injury and peritonitis in a traumatic rabbit model was increasingly severe in group 4. At T2, the temperature was significantly decreased, and the lactate was signif i cantly increased in group 4; these results were significantly different from those in the other groups. Hypothermia and acidosis are important factors that affect clotting factor and platelet activity; as temperature decreases and acidosis is aggravated, the incidence of coagulopathy will increase. Clotting factor activity and platelet aggregation may be reduced by hypothermia (<33–34 °C).[11,23]White et al[24]found that acidosis may reduce the activity of the factor Xa/Va complex, and the activity will be persistently reduced by the increasing acidosis severity. NEUT% values were significantly increased in group 4, implying that group 4 had inflammation. Although not fully elucidated, there is evidence that these values indicate cross-talk between inflammation and coagulation. Tissue factor, the thrombin-thrombomodulin protein C pathway, endothelium, complement proteins, proinf l ammatory cytokines, and platelets play important roles in this interaction between inflammation and coagulation.[23,25–27]The inflammation cytokines enhance endothelial expression/release of thrombomodulin resulting in enhanced activation of PC.[28]With increasingly severe trauma, the excessive increases in plasma catecholamine increase the release of tPA and PC, and the excessive consumption of coagulation factors and platelets, resulting in predominant hypocoagulability.[29]With increasing trauma severity (as measured by ISS), the coagulation monitored by TEG changes from normal (minor tissue injuries) to hypercoagulable (ISS 10–25), to hypocoagulable (ISS 20–35) and hyperf i brinolytic (ISS >35).[1]
We observed that at T2, coagulation differed prominently among the three trauma groups (2, 3, 4), even though the platelets count in the trauma groups (2, 3, and 4) were significantly decreased compared with that in the sham group and were not different among the groups. The results suggest that platelet count was not the key factor in coagulopathy, although one of the current debates on traumatic coagulopathy is the decrease or consumption of platelets. Early platelet dysfunction might be positively correlated with the severity of coagulopathy. In a clinical study, Ostrowski et al[30]found that trauma-induced platelet excessive activation may lower platelet responsiveness or hemostatic potential, so the remaining platelets cannot adequately support clot formation. In addition, adenosine diphosphate (ADP) is one of the main activators of platelet aggregation, but traumatic shock-induced acidosis causes excess ADP generation, which also lowers platelet responsiveness.[31]
There are several limitations in this study. First, rabbits have a different coagulation system from humans, which may have impacted our results. Second, we induced a multiple injuries model focused on abdominal trauma, and the observation period ended 4 hours posttrauma (T2) or rabbits death; the impact of injury to different regions of the body and the later effect of trauma on coagulopathy was not considered in the present study. Third, with the limitation of experimental conditions, many impact factors regarding traumatic coagulopathy were not assayed such as pH, base excess and PaO2. Lactate is the earliest marker seen in blood during hypoxia and oxidative stress, so we used lactate to reflect the severity of hypoxia. Fourth, traditionalcoagulation tests [prothrombin time (PT), activated partial thromboplatin time (APTT), international normalized ratio (INR)] end once fibrin formation starts; thus, these assays are only sensitive to identify hypocoagulation. Therefore, we only used TEG to evaluate coagulation.
CONCLUSION
The present study suggests that shock combined with different abdominal traumas causes diverse coagulopathies in the early phase. Nontraumatic hemorrhagic shock shows no obvious effect on blood coagulation, hemorrhagic shock combined with blunt hepatic injury shows hypercoagulability, and hemorrhagic shock with hepatic blunt-impact injury coupled with peritonitis caused by intestinal rupture demonstrates a tendency toward hypocoagulability.
ACKNOWLEDGMENTS
This research was supported by Guangxi Natural Science Foundation (No. 2015GXNSFAA139195) and Guangxi Emergency Medicine and Medical Rescue Talent Upland Foundation (No. GXJZ201403). The manuscript listed below was edited for proper English language, grammar, punctuation, spelling, and overall style by one or more of the highly qualif i ed native English speaking editors at American Journal Experts.
Funding: This study was funded by Guangxi Natural Science Foundation (No. 2015GXNSFAA139195) and Guangxi Emergency Medicine and Medical Rescue Talent Upland Foundation (No. GXJZ201403).
Ethical approval: This study was approved by the Institutional Ethics Committee of Guangxi Medical University.
Conflicts of interest: The authors declare that there are no conf l icts of interest relevant to the content of the article.
Contributors: Wu R proposed the study and wrote the fi rst draft. All authors read and approved the fi nal version of the paper.
REFERENCES
1 Johansson PI. Coagulation monitoring of the bleeding traumatized patient. Curr Opin Anaesthesiol. 2012;25(2):235–41.
2 Johansson PI, Ostrowski SR, Secher NH. Management of major blood loss: an update. Acta Anaesthesiol Scand. 2010;54(9):1039–49.
3 Brohi K, Cohen MJ, Davenport RA. Acute coagulopathy of trauma: mechanism, identification and effect. Curr Opin Crit Care. 2007;13(6):680–5.
4 Engels PT, Rezende-Neto JB, Al Mahroos M, Scarpelini S, Rizoli SB, Tien HC. The natural history of trauma-related coagulopathy: implications for treatment. J Trauma. 2011;71(5 Suppl 1):S448–55.
5 Wafaisade A, Wutzler S, Lefering R, Tjardes T, Banerjee M, Paffrath T, et al. Trauma Registry of DGU. Drivers of acute coagulopathy after severe trauma: a multivariate analysis of 1987 patients. Emerg Med J. 2010;27(12):934–9.
6 Loveland JA, Boffard KD. Damage control in the abdomen and beyond. Br J Surg. 2004;91(9):1095–1101.
7 Burggraf M, Payas A, Kauther MD, Schoeneberg C, Lendemans S. Evaluation of clotting factor activities early after severe multiple trauma and their correlation with coagulation tests and clinical data. World J Emerg Surg. 2015;10:43.
8 Dobson GP, Letson HL, Sharma R, Sheppard FR, Cap AP. Mechanisms of early trauma-induced coagulopathy: The clot thickens or not? J Trauma Acute Care Surg. 2015;79(2):301–9.
9 Moore EE, Cogbill TH, Jurkovich GJ, Shackford SR, Malangoni MA, Champion HR. Organ injury scanling: spleen and liver (1994 revision). J Trauma. 1995;38(3):323–4.
10 Lee SK, Carrillo EH, Rosenthal A, Sanchez R, Kiffin C, Davare DL. Acute Care/Trauma Surgeon's role in obstetrical/ gynecologic emergencies (The OBCAT Alert). World J Emerg Med. 2016;7(4):274–7.
11 Frith D, Brohi K. The acute coagulopathy of trauma shock: clinical relevance. Surgeon. 2010;8(3):159–63.
12 Schöchl H, Cadamuro J, Seidl S, Franz A, Solomon C, Schlimp CJ, et al. Hyperfibrinolysis is common in out-ofhospital cardiac arrest: results from a prospective observational thromboelastometry study. Resuscitation. 2013;84(4):454–9.
13 Boura C, Caron A, Longrois D, Mertes PM, Labrude P, Menu P. Volume expansion with modif i ed hemoglobin solution, colloids, or crystalloid after hemorrhagic shock in rabbits: effects in skeletal muscle oxygen pressure and use versus arterial blood velocity and resistance. Shock. 2003;19(2):176–82.
14 Lawton LD, Roncal S, Leonard E, Stack A, Dinh MM, Byrne CM, et al. The utility of Advanced Trauma Life Support (ATLS) clinical shock grading in assessment of trauma. Emerg Med J. 2014;31(5):384–9.
15 Fung YL, Tung JP, Foley SR, Simonova G, Thom O, Staib A, et al. Stored blood transfusion induces transient pulmonary arterial hypertension without impairing coagulation in an ovine model of nontraumatic haemorrhage. Vox Sang. 2013;105(2):150–8.
16 Brohi K, Cohen MJ, Ganter MT, Matthay MA, Mackersie RC, Pittet JF. Acute traumatic coagulopathy: initiated by hypoperfusion: modulated through the protein C pathway? Ann Surg. 2007;245(5):812–8.
17 Frith D, Goslings JC, Gaarder C, Maegele M, Cohen MJ, Allard S, et al. Def i nition and drivers of acute traumatic coagulopathy: clinical and experimental investigations. J Thromb Haemost. 2010;8(9):1919–25.
18 Mulier KE, Greenberg JG, Beilman GJ. Hypercoagulability in porcine hemorrhagic shock is present early after trauma and resuscitation. J Surg Res. 2012;174(1):e31–5.
19 Ding H, Cao XY, Ma XG, Zhou WJ. Endothelial cell injury with inflammatory cytokine and coagulation in patients with sepsis. World J Emerg Med. 2013;4(4):285–9.
20 Park MS, Martini WZ, Dubick MA, Salinas J, Butenas S, Kheirabadi BS, et al. Thromboelastography as a better indicator of hypercoagulable state after injury than prothrombin time or activated partial thromboplastin time. J Trauma. 2009;67(2):266–75.
21 Martini J, Cabrales P, Fries D, Intaglietta M, Tsai AG. Effects of fibrinogen concentrate after shock/resuscitation: a comparison between in vivo microvascular clot formation and thromboelastometry*. Crit Care Med. 2013;41(11):e301–308.
22 Duan K, Yu W, Lin Z, Tan S, Bai X, Xu L, et al. A time course study of acute traumatic coagulopathy prior to resuscitation: from hypercoagulation to hypocoagulation caused by hypoperfusion? Transfus Apher Sci. 2014;50(3):399–406.
23 Ganter MT, Pittet JF. New insights into acute coagulopathy in trauma patients. Best Pract Res Clin Anaesthesiol. 2010;24(1):15–25.
24 White NJ, Martin EJ, Brophy DF, Ward KR. Coagulopathy and traumatic shock: characterizing hemostatic function during the critical period prior to fluid resuscitation. Resuscitation. 2010;81(1):111–6.
25 Brohi K. Trauma induced coagulopathy. J R Army Med Corps. 2009;155(4):320–2.
26 van der Poll T, Boer JD, Levi M. The effect of inflammation on coagulation and vice versa. Curr Opin Infect Dis. 2011;24(3):273–8.
27 Gao JP, Ying KJ. Thrombolysis during extended cardiopulmonary resuscitation for autoimmune-related pulmonary embolism. World J Emerg Med. 2015;6(2):153–6.
28 Brohi K, Cohen MJ, Ganter MT, Schultz MJ, Levi M, Mackersie RC, et al. Acute coagulopathy of trauma: hypoperfusion induces systemic anticoagulation and hyperfibrinolysis. J Trauma. 2008;64(5):1211–7.
29 Hess JR, Brohi K, Dutton RP, Hauser CJ, Holcomb JB, Kluger Y, et al. The coagulopathy of trauma: a review of mechanisms. J Trauma. 2008;65(4):748–54.
30 Ostrowski SR, Sorensen AM, Larsen CF, Johansson PI. Thrombelastography and biomarker profiles in acute coagulopathy of trauma: a prospective study. Scand J Trauma Resusc Emerg Med. 2011;19:64.
31 Carroll RC, Craft RM, Langdon RJ, Clanton CR, Snider CC, Wellons DD, et al. Early evaluation of acute traumatic coagulopathy by thromboelastography. Transl Res. 2009;154(1):34–9.
Received October 20, 2016
Accepted after revision March 6, 2017
World J Emerg Med 2017;8(2):141–147
10.5847/wjem.j.1920–8642.2017.02.011
Hui-min Zhao, Email: hmzhao2006@163.com
 World journal of emergency medicine2017年2期
World journal of emergency medicine2017年2期
- World journal of emergency medicine的其它文章
- Instructions for Authors
- Information for Readers
- World Journal of Emergency Medicine
- Acute airway emergency caused by an organic foreign body located in the laryngeal mucosa
- Bacteremia or pseudobacteremia? Review of pseudomonas fl uorescens infections
- An unusual case of renal calculi leading to myocardial infarction and cardiogenic shock