
通过式固相萃取净化/液相色谱-串联质谱法快速检测水产品中11种青霉素残留
2017-04-08 03:51:03郭萌萌李兆新潘明轩吴海燕邢丽红孙晓杰
分析测试学报 2017年3期
郭萌萌,李兆新*,王 智,潘明轩,3,吴海燕,邢丽红,孙晓杰
(1.中国水产科学研究院 黄海水产研究所 农业部水产品质量安全检测与评价重点实验室,山东 青岛 266071;2.青岛市产品质量监督检验研究院 青岛市产品质量检验技术研究所,山东 青岛 266061;3.上海海洋大学,上海 201306)
通过式固相萃取净化/液相色谱-串联质谱法快速检测水产品中11种青霉素残留
郭萌萌1,李兆新1*,王 智2,潘明轩1,3,吴海燕1,邢丽红1,孙晓杰1
(1.中国水产科学研究院 黄海水产研究所 农业部水产品质量安全检测与评价重点实验室,山东 青岛 266071;2.青岛市产品质量监督检验研究院 青岛市产品质量检验技术研究所,山东 青岛 266061;3.上海海洋大学,上海 201306)
采用通过式固相萃取净化策略去除样品基质中的脂肪和磷脂等杂质干扰,结合液相色谱-串联质谱检测,建立了水产品中11种青霉素残留的同时快速分析方法。样品经80%乙腈水溶液提取,Oasis PRiME HLB通过式固相萃取柱净化,C18色谱柱分离,0.05%甲酸乙腈溶液和0.05%甲酸水溶液梯度洗脱,多反应监测正离子模式扫描,内标法定量。11种目标物在相应浓度范围内线性关系良好,相关系数不低于0.99,检出限为0.30~1.5 μg/kg。基质加标回收率为85.5%~110%,相对标准偏差(RSD)为5.9%~14.3%。该方法前处理操作简便,灵敏度和准确度高,可实现水产品中多种青霉素药物残留的同时快速测定。
青霉素;通过式固相萃取;液相色谱-串联质谱(LC-MS/MS);水产品
青霉素类药物属β-内酰胺抗生素,为预防和治疗细菌感染的首选抗生素,是临床最常用的抗生素之一,也被广泛应用于畜牧、水产等养殖业。在水产养殖中,有些不法养殖业者在饲料中违规添加青霉素类药物或用药不规范将导致药物在水产品中残留,不仅对消费者造成直接的健康危害,还增加了人畜共患细菌耐药菌株泛滥的可能。许多国家和地区对青霉素类药物在动物源性食品中的残留进行了严格限制,我国农业部235号公告[1]规定动物肌肉中阿莫西林、氨苄西林、青霉素G、氯唑西林、苯唑西林的最高残留限量分别为50,50,50,300,300 μg/kg;欧盟指令No.2377/90/EEC[2]规定动物肌肉中阿莫西林、氨苄西林、青霉素G、苯唑西林、氯唑西林、双氯西林、萘夫西林的最高残留限量分别为50,50,50,300,300,300,300 μg/kg。
青霉素类药物多残留的分析方法以高效液相色谱法(HPLC)[3-4]和液相色谱-串联质谱法(LC-MS/MS)[5-8]为主,其中LC-MS/MS法因定性能力更强而成为目前国际上广泛采用的青霉素残留分析方法。现有方法多采用固相萃取(SPE)技术净化样品,步骤繁琐,且部分化合物的回收率不高,如Macarov等[9]采用ENV+Isolute固相萃取柱净化结合LC-MS/MS分析方法实现了畜禽肉中9种青霉素的多残留分析;Junza等[10]采用HLB和Strata固相萃取柱净化结合UPLC-MS/MS分析方法测定了牛奶中7种青霉素残留,以上方法均以哌拉西林为内标进行内标法校正,但阿莫西林的回收率仅为50%左右;也有研究采用基质固相分散萃取[11]和分散固相萃取[12]净化,使用LC-MS/MS法检测猪肉或水产品中的8~12种青霉素,简化了样品前处理方法,以青霉素G-d7为内标,采用内标法定量获得了较好的回收率(>70%)。
1 实验部分
1.1 仪器、试剂与材料
Prominence UFLC超快速液相色谱(日本Shimadzu公司);5500 QTRAP四极杆-线性离子阱复合质谱检测系统(美国AB SCIEX公司);T18 Basic均质机(德国IKA公司);XW-80A旋涡混合器(上海医大仪器厂);Himac CR 22GⅡ高速离心机(日本Hitachi公司);N-EVAP112氮吹仪(美国Organomation公司);Milli-Q超纯水仪(美国Millipore公司)。
标准品:阿莫西林、氨苄西林、青霉素G、青霉素V、苯唑西林、氯唑西林、萘夫西林、双氯西林、哌拉西林(德国Dr.Ehrensorfer公司);阿洛西林、甲氧西林、阿莫西林-d4、氨苄西林-d5、青霉素G-d7、萘夫西林-d6(加拿大Toronto Research Chemicals公司);乙腈(HPLC级,美国Merck公司);甲酸(HPLC级,美国Fluka公司);超纯水(18.2 MΩ·cm);Oasis PRiME HLB固相萃取柱(200 mg/6 mL,美国Waters公司),其他未作特殊说明的试剂均为分析纯。
1.2 样品预处理
准确称取2.5 g匀浆试样于15 mL聚丙烯离心管中,加入内标物(各10 ng),静置10 min后加入9 mL 80%乙腈水溶液,涡旋混合1 min,超声提取10 min,在4 ℃下10 000 r/min离心5 min,移取上清液至10 mL比色管中,再用 80%乙腈水溶液定容至10 mL。
取约1 mL上述提取液润洗Oasis PRiME HLB固相萃取柱,自然流速流干,弃去,再准确移取2.0 mL提取液加载至SPE小柱上,保持流速为1滴/s,收集流出液。将流出液于35 ℃氮气吹至略低于0.5 mL,用水定容至0.5 mL,超滤管以12 000 r/min高速离心10 min,供LC-MS/MS测试。
1.3 仪器分析条件
液相色谱条件:Kinetex XB-C18色谱柱(2.1 mm×100 mm,2.6 μm);柱温:35 ℃;流速:0.40 mL/min;进样量:10 μL;流动相:A为0.05%的甲酸水溶液,B为0.05%的甲酸乙腈溶液;洗脱梯度:0~1.0 min,2% B;1.0~3.0 min,2%~50% B;3.0~6.0 min,50%~100%B;6.0~8.0 min,100%B;8.1~10.0 min,2%B。
第四,落实防疫成本费,稳定防疫团队。畜禽动物防疫工作费用需要财政预算颁布,费用有工作人员薪资、免疫药物费用、疫苗费用等。这一方面稳定防疫团队,一方面减轻防疫团队和养殖户的负担。完善动物防疫系统,让各级乡、村均有专业防疫人员,让动物防疫工作真正落实到各个角落,从而做到“无不漏”防疫原则。
质谱条件:电喷雾离子源(ESI),多反应监测(MRM),正离子模式;喷雾电压:5.5 kV;气帘气压力:0.24 MPa;碰撞气压力:0.02 MPa;温度500 ℃;碰撞室入口电压:10 V;碰撞室出口电压:12 V;驻留时间:50 ms;离子源Gas1:0.34 MPa;Gas2:0.34 MPa。其它参数见表1。
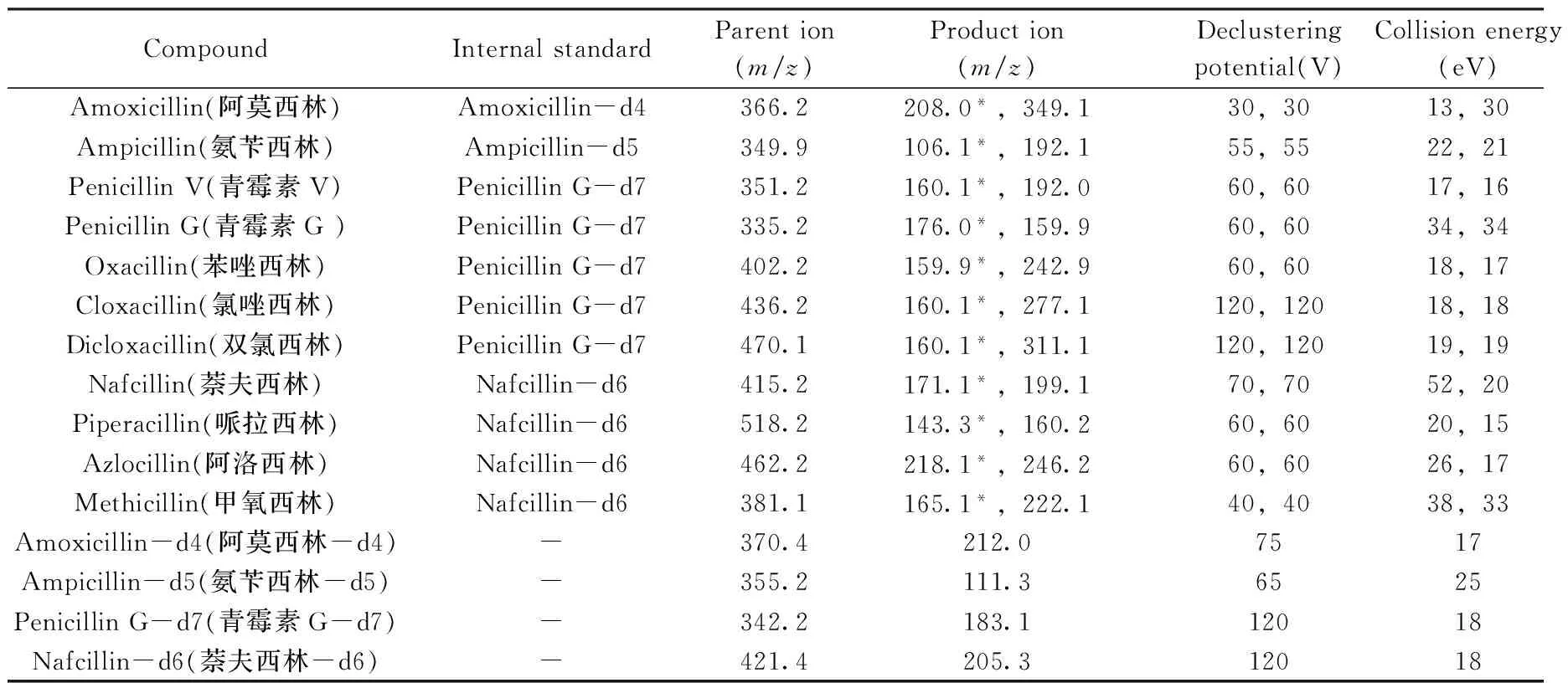
表1 11种目标物的质谱分析参数Table 1 Mass spectrometric parameters for 11 target compounds
*quantitative ion
2 结果与讨论
2.1 色谱条件的优化
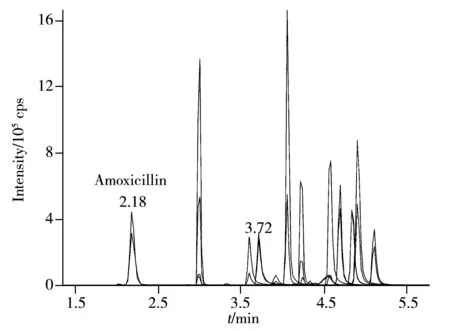
图1 11种青霉素混合标准溶液的总离子流色谱图(10.0 μg/L)Fig.1 Total ion chromatogram of 11 penicillins mixed standard solution(10.0 μg/L)mobile phase:acetonitrile(0.005% formic acid)-water(0.005% formic acid)
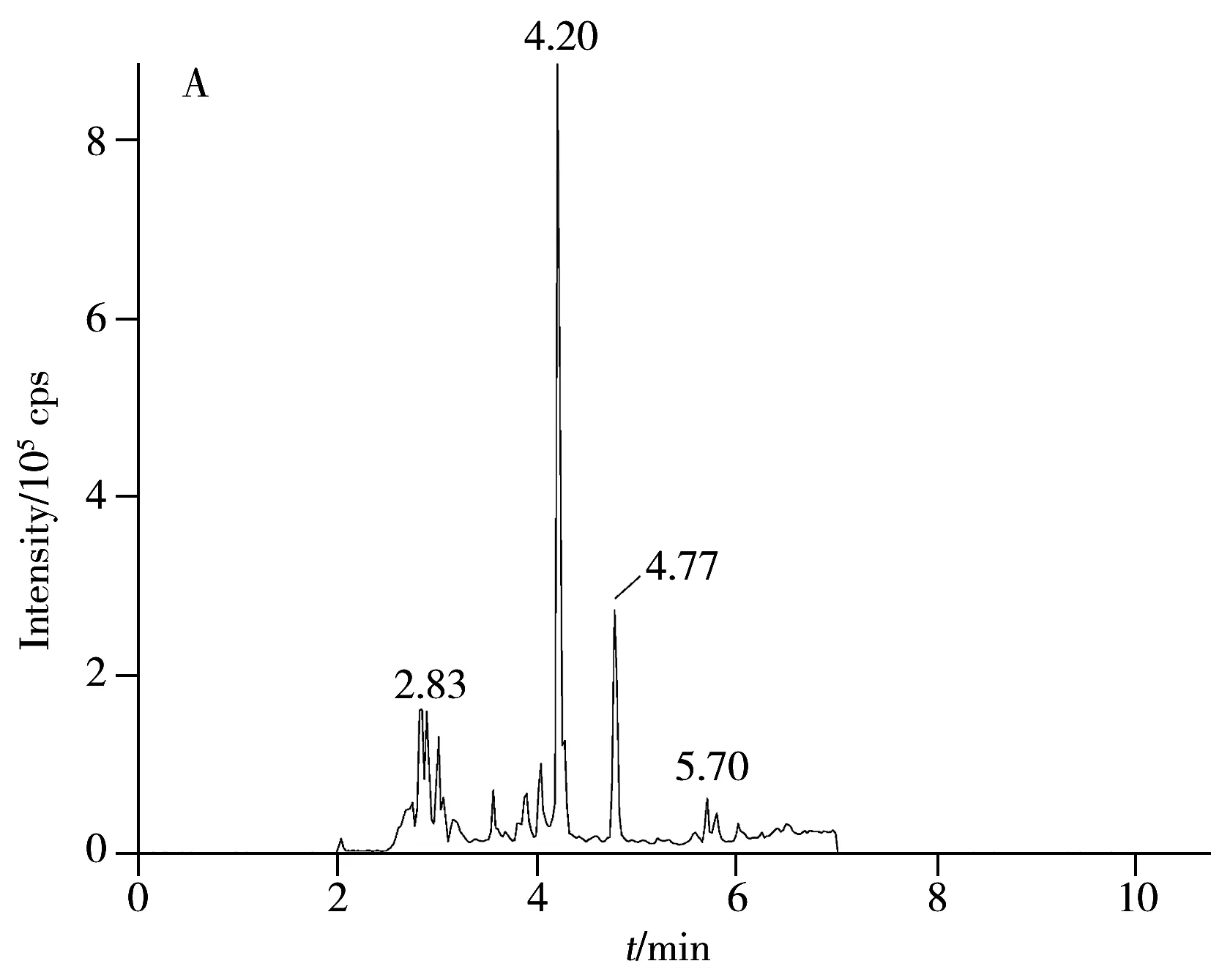
考虑到青霉素药物化学结构中含有羧基,而甲醇的化学结构含有羟基,会加速青霉素降解为青霉噻唑酸酯,因此本方法选择乙腈-水作为流动相体系。已有关于青霉素检测方法的文献[9-13]多采用乙腈(0.1%甲酸)-水(0.1%甲酸)进行梯度洗脱,可获得较好的分离效果和色谱峰形,也有文献报道[14]采用极少量的甲酸(0.005%)能获得更高的灵敏度。本研究进一步优化了乙腈-水流动相体系中不同甲酸浓度(0.005%,0.02%,0.05%,0.1%)对青霉素药物色谱分离的影响。实验发现甲酸添加浓度为0.005%时,各化合物的响应值较高、分离度较好,仅阿莫西林出峰较快、峰形较宽(图1),且在样品溶液中无法与杂质分离(图2A);随着甲酸浓度增加至0.1%时,阿莫西林的保留能力增强,峰形变窄、响应值增强,但出峰较慢的双氯西林等其他化合物的保留能力减弱,影响了色谱分离效果。兼顾上述影响因素,本方法最终选择在乙腈-水体系中各添加0.05%的甲酸作为流动相,不仅获得了较佳的色谱峰形、分离效果和灵敏度,且在复杂基质溶液中实现了11种青霉素药物残留与杂质的良好分离(以阿莫西林为例,见图2B)。
2.2 样品前处理方法的优化
2.2.1 提取溶剂的选择 青霉素类药物易溶于水,且分子结构中含β-内酰胺环,易受酸碱、重金属、羟胺基等影响发生环的裂解而稳定性较差。本方法兼顾青霉素类药物的溶解性和稳定性,选取乙腈水溶液为提取溶剂,同时分别考察了不同浓度(70%,80%,90%)乙腈水溶液对目标物的提取效果。结果表明,以平均回收率计算,其提取率的大小顺序为80%乙腈水溶液(~90%)>90%乙腈水溶液(~83%)>70%乙腈水溶液(~70%),与文献报道的80%乙腈水溶液对药物残留的萃取率较高的实验结果一致[15],可能原因是水产品含丰富的蛋白质,80%乙腈水溶液可使蛋白质发生缓慢且彻底的变性,释放基质中的药物残留,而小于80%的乙腈不能引起蛋白质的完全变性,高于80%的乙腈使蛋白质迅速凝聚而包裹药物导致萃取率偏低。因此本方法选用80%乙腈水溶液为提取剂。

2.2.2 样品净化方法的优化 脂肪和磷脂类化合物是水产品基质中药物残留分析的主要干扰因素,这些物质不仅干扰LC-MS/MS分析,还会影响色谱柱固定相的活性位点并降低其分辨率[16],甚至污染质谱仪。本研究为提高方法的准确度和延长色谱柱、质谱仪的使用寿命,拟增加快速、有效的净化除脂步骤。采用通过式固相萃取净化策略,以Oasis PRiME HLB固相萃取柱净化样品,上样前先用约1 mL样品溶液润洗小柱,然后直接将样品溶液加载至SPE柱以吸附脂肪和磷脂等杂质,整个净化过程不损失目标组分,不仅有效净化了样品溶液,显著提高了前处理效率,还增加了目标物和样品的分析通量。通过比较Oasis PRiME HLB柱净化前后空白鱼肉样品的全扫描总离子流图(图3),可以看出,Oasis PRiME HLB净化后的鱼肉样品,以基质成分的强度估算,基质成分明显减少。
本实验还考察了上样量对目标物回收率的影响。分别选取1,2,3,4 mL不同的上样体积,通过比较平均回收率优化最佳的上样量。实验结果表明,上样体积为1 mL和2 mL时,青霉素药物的平均回收率高于90%且样品净化溶液较澄清。从方法的灵敏度考虑,本方法选取2 mL为最佳上样体积。
2.2.3 样品浓缩条件的优化 采用氮气吹扫的方式对样品溶液进行浓缩,因样品溶液含水量约为20%,实验发现乙腈挥发后,少量的水溶液较难挥发,使得氮吹浓缩时间延长,氮吹至干时部分目标物的回收率不足60%。因此,本方法浓缩样品溶液时,氮吹至略小于0.5 mL即取出定容,不仅缩短了浓缩时间,同时也保证了较高的回收率。
2.3 基质效应的评价
为评估本方法的基质效应,实验考察了用青霉素混合标准系列溶液校正和基质标准系列溶液校正对目标物测定结果的影响,移取10 μg/L的青霉素混合标准溶液0.5 mL,于35 ℃氮气吹干,加入按“1.2”步骤制备的空白基质溶液溶解并定容至0.5 mL,此时样品溶液中各目标物的含量为10 μg/kg,作为考察定量结果的理论值,实验结果见表2。结果表明,本方法经有效的前处理净化及内标法校正,用青霉素标准系列溶液校正的定量结果与用基质标准系列溶液校正的定量结果基本一致,均与理论值吻合较好,无明显基质效应。
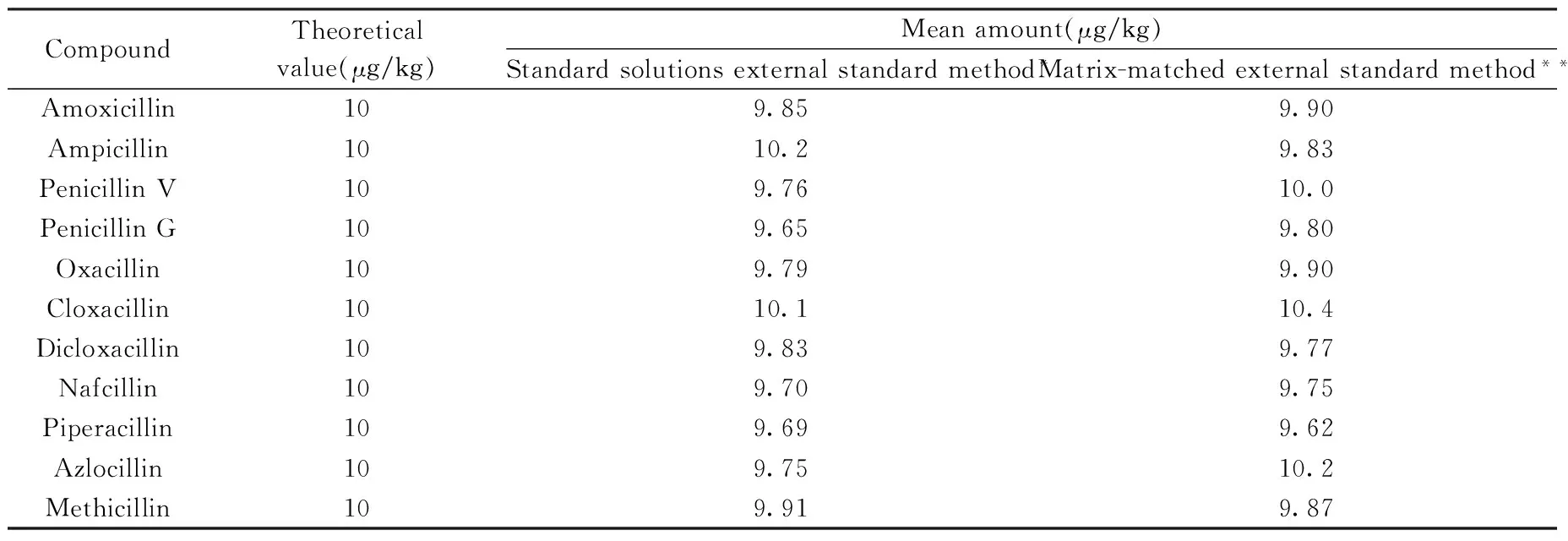
表2 不同校正方法对目标物定量结果的影响(n=3)Table 2 Quantitative results of 11 target compounds by different calibration methods(n=3)
*the calibration curves were prepared using penicillins standard solutions;**the calculation curves were prepared using matrix-matched standard solution
2.4 灵敏度、准确度与精密度
取适量青霉素药物混合标准溶液和内标溶液,配制一系列浓度梯度的标准溶液,在“1.3”条件下依次测定,以各组分和内标物的峰面积比值为纵坐标(y),各组分的质量浓度为横坐标(x,μg/L)进行线性回归分析。在空白样品中添加低浓度的标准溶液,按“1.2”步骤进行样品前处理后进样测定,以信噪比S/N≥3和S/N≥10分别确定检出限(LOD)及定量下限(LOQ)。11种目标物的线性范围、回归方程、相关系数、检出限及定量下限见表3。结果表明,11种青霉素的线性关系良好,检出限为0.30~1.5 μg/kg,方法的灵敏度适用于多组分青霉素药物残留的高灵敏定量分析。
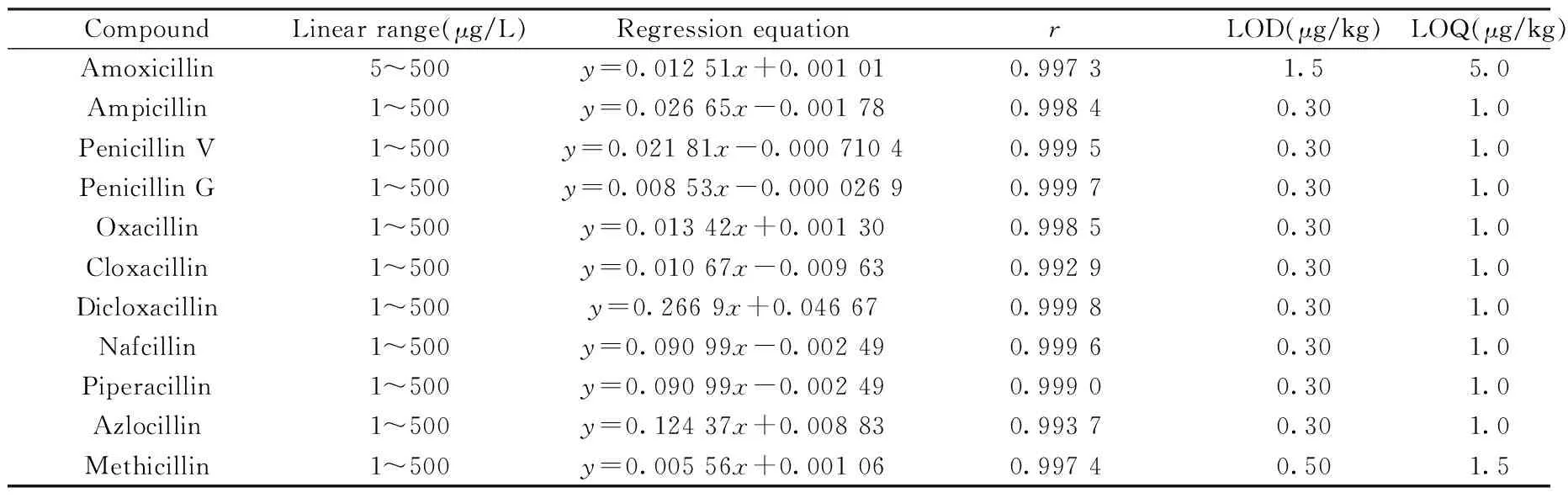
表3 11种青霉素的线性范围、回归方程、相关系数、检出限及定量下限Table 3 Linear ranges,regression equations,correlation coefficients,limits of detection and quantitation of 11 penicillins
选取阴性鲤鱼、甲鱼和虾的肌肉为测试基质,分别添加相当于5,50,300 μg/kg浓度的混合标准溶液进行测定,每个浓度设6个平行。结果显示,11种目标物在鲤鱼肌肉中的加标回收率为85.5%~105%,相对标准偏差为5.9%~12.1%;在甲鱼肌肉中的加标回收率为87.8%~110%,相对标准偏差为7.6%~14.3%;在虾肌肉中的加标回收率为89.0%~102%,相对标准偏差为7.7%~13.0%。以上结果说明,该方法的准确度高,精密度好,可满足药物残留的日常监测要求。
2.5 实际样品的测定
应用本方法检测了30个水产品(鲤鱼、甲鱼、对虾、鳗鱼等)的青霉素药物残留情况,其中1个鳗鱼样品检出青霉素G,含量为3.75 μg/kg,远低于我国动物肌肉中标准限量50 μg/kg,其他青霉素药物均未检出。
3 结 论
本文利用通过式Oasis PRiME HLB固相萃取净化策略,省去了常规固相萃取净化方法的活化和淋洗步骤,采用直接上样吸附杂质成分,简化了样品前处理步骤,且不损失目标组分,实现了11种青霉素药物的同步提取;同时采用同位素内标法定量,解决了大多数青霉素残留方法中阿莫西林回收率不高的难题,提高了青霉素多残留测定方法的准确度。该方法的建立为同时分析生物基质中多种青霉素药物残留提供了新的技术手段。
[1] Ministry of Agriculture.No.235 Bulletin of the Ministry of Agriculture of the People’s Republic of China(农业部.中华人民共和国农业部公告第235号).[2002-12-24].http://www.moa.gov.cn/zwllm/nybz/index_2.htm.
[2] Council Regulation(EEC) No 2337/90 of 26 June 1990 Laying Dowm a Community Procedure for the Establishment of Maximum Residue Limits of Veterinary Medicinal Products in Foodstuffs of Animal Origin.Official Journal of the European Communities,1997,L67:1.
[3] GB 29682-2013.Multi Residue Determination of the Penicillins in Aquatic Products by High Performance Liquid Chromatographic Method.National Food Safety Standards(水产品中青霉素类药物多残留的测定 高效液相色谱法.食品安全国家标准).
[4] Ye N S,Gu X X,Zhang Q.Chin.J.Anal.Lab.(叶能胜,谷学新,张琦.分析试验室),2010,29(6):73-77.
[5] Dorival-García N,Junza A,Zafra-Gómez A,Barrón D,Navalón A.FoodControl,2016,60:382-393.
[6] Chico J,Rúbies A,Centrich F,Companyó R,Prat M D,Granados M.J.Chromatogr.A,2008,1213(2):189-199.
[7] GB/T 22952-2008.Determination of Amoxicillin,Ampicillin,Piperacillin,Penicillin G,Penicillin V,Oxacillin,Cloxacillin,Nafcillin,Dicloxacillin Residues in Fugu and Eel——LC-MS/MS Method.National Standards of the People’s Republic of China(河豚鱼和鳗鱼中阿莫西林、氨苄西林、哌拉西林、青霉素G、青霉素V、苯唑西林、氯唑西林、萘夫西林、双氯西林残留量的测定 液相色谱-串联质谱法.中华人民共和国国家标准).
[8] GB/T 21315-2007.Determination of Penicillins Residues in Foodstuffs of Animal Origin—LC-MS/MS Method.National Standards of the People’s Republic of China(动物源性食品中青霉素族抗生素残留量检测方法 液相色谱-质谱/质谱法.中华人民共和国国家标准).
[9] Macarov C A,Tong L,Martínez-Huélamo M,Hermo M P,Chirila E,Wang Y X,Barrón D,Barbosa J.FoodChem.,2012,135:2612-2621.
[10] Junza A,Amatya R,Barrón D,Barbosa J.J.Chromatogr.B,2011,879:2601-2610.
[11] Huang Z,Pan X D,Huang B F,Xu J J,Wang L M,Ren Y P.FoodControl,2016,66:145-150.
[12] Guo M M,Li Z X,Tan Z J,Wu H Y,Han X Q,Leng K L,Zhai Y X.J.Instrum.Anal.(郭萌萌,李兆新,谭志军,吴海燕,韩现芹,冷凯良,翟毓秀.分析测试学报),2011,30(9):969-975.
[13] Liu Y,Zhu K,Wang J F,Huang X Y,Wang G L,Li C Y,Cao J,Ding S Y.J.Chromatogr.B,2016,1008:74-80.
[14] Becker M,Zittlau E,Petz M.Anal.Chim.Acta,2004,520:19-32.
[15] Jia F Y,Wang W W,Liu Z B,Yin J G,Liu Y M.ActaChim.Sin.(贾风燕,王文文,刘振波,殷军港,刘永明.化学学报),2012,70(4):485-491.
[16] Pensado L,Casais M C,Mejuto M C,Cela R.J.Chromatogr.A,2005,1077(2):103-109.
Rapid and Simultaneous Determination of 11 Penicillin Residues in Fishery Products by Pass-through SPE Purification and Liquid Chromatography-Tandem Mass Spectrometry
GUO Meng-meng1,LI Zhao-xin1*,WANG Zhi2,PAN Ming-xuan1,3,WU Hai-yan1,XING Li-hong1,SUN Xiao-jie1
(1.Key Laboratory of Testing and Evaluation for Aquatic Product Safety and Quality,Ministry of Agriculture,Yellow Sea Fisheries Research Institute,Chinese Academy of Fishery Sciences,Qingdao 266071,China;2.Qingdao Institute of Product Quality Inspection and Technical Research,Qingdao Product Quality Supervision and Testing Research,Qingdao 266061,China;3.Shanghai Ocean University,Shanghai 201306,China)
A method was developed for the rapid and simultaneous determination of 11 penicillins in fishery products by pass-through SPE purification with liquid chromatography tandem mass spectrometry(LC-MS/MS).The samples were extracted with acetonitrile-water(80∶20,by volume),and then cleaned up with an Oasis PRiME HLB pass-through SPE column.The separation of 11 penicillins were performed on a Kinetex XB-C18(2.1 mm×100 mm,2.6 μm) column by using a mixture of 0.05% formic acid and acetonitrile containing 0.05% formic acid as mobile phase.Qualitative and quantitative analyses of the analyte were carried out under the multiple reaction monitoring(MRM) mode with positive electrospray ionization.And the isotope internal standards were employed for the quantification.The calibration curves were linear well in corresponding concentration ranges,with correlation coefficient over 0.99.The detection limits ranged from 0.30 μg/kg to 1.5 μg/kg.The average spiked recoveries for 11 penicillins were between 85.5%and 110% with relative standard deviations(RSDs) of 5.9%-14.3%.The proposed method is accurate,sensitive and high-efficient,and is practical for the determination of penicillins in fishery products.
penicillins;pass-through solid-phase extraction;liquid chromatography tandem mass spectrometry(LC-MS/MS);fishery products
10.3969/j.issn.1004-4957.2017.03.007
2016-10-10;
2016-11-20
农业行业标准修订项目(2015-439)
O657.63;TQ465.1
A
1004-4957(2017)03-0337-06
*通讯作者:李兆新,博士,研究员,研究方向:水产品安全与质量控制,Tel:0532-85836348,E-mail:lizx@ysfri.ac.cn
猜你喜欢
课堂内外·初中版(科学少年)(2024年6期)2024-07-08 17:00:28
Chinese Physics B(2022年5期)2022-05-16 07:09:10
东坡赤壁诗词(2020年3期)2020-07-04 02:50:05
祝您健康·文摘版(2019年10期)2019-10-14 02:07:23
启迪与智慧·下旬刊(2019年2期)2019-09-10 07:22:44
中国蜂业(2018年4期)2018-05-09 06:25:08
文理导航·科普童话(2016年7期)2017-02-04 20:21:18
新丝路(下旬)(2016年10期)2016-06-05 15:22:12
当代化工研究(2016年6期)2016-03-20 16:21:46
新疆农垦科技(2014年6期)2014-02-28 19:20:19
