
固相萃取/超高效液相色谱-串联质谱法测定畜肉中16种镇静剂类兽药残留
2017-04-08 03:12刘家阳贾宏新
分析测试学报 2017年3期
刘家阳,黄 旭,贾宏新
(辽宁省食品检验检测院 ,辽宁 沈阳 110015)
固相萃取/超高效液相色谱-串联质谱法测定畜肉中16种镇静剂类兽药残留
刘家阳1,黄 旭2,贾宏新3*
(辽宁省食品检验检测院 ,辽宁 沈阳 110015)
建立了畜肉中16种镇静剂类兽药残留的固相萃取净化/超高效液相色谱-串联质谱(LC-MS/MS)的同时测定方法。样品均质后经氢氧化钠溶液水解,加入盐酸溶液提取。提取液经固相萃取柱MCX净化,洗脱液氮气吹干后用流动相溶解,经0.22 μm滤膜过滤,采用Waters ACQUITY UPLC BEH C18(2.1 mm×100 mm,1.7 μm)色谱柱分离,在电喷雾电离源(ESI)和多反应监测(MRM)正离子模式下测定,外标法定量。结果表明:采用基质匹配外标法测定,16种镇静剂类化合物在一定浓度范围内呈良好的线性关系(r2≥0.996 8),在样品中的检出限和定量下限分别为0.01~0.05 μg/kg和0.1~0.5 μg/kg,在3个浓度加标水平下的平均回收率为71.6%~112%,相对标准偏差(RSD)为2.9%~15.8%。市售多种鲜肉制品的测定结果表明,该方法选择性好、操作简单快速、结果准确,适用于畜肉中16种镇静剂类兽药残留的同时快速测定。
固相萃取;超高效液相色谱-串联质谱;镇静剂;畜肉
近年来畜肉及其制品中兽药残留问题逐渐成为人们关注的焦点[1]。镇静剂药物是一类通过抑制中枢神经系统减弱其生理机能而达到缓和激动、消除躁动、恢复安静情绪的药物[2]。近年来,安眠镇静类药物被个别不法畜牧业主作为生长促进剂用于养殖业[3]。镇静剂类药物在畜禽体内具有一定的代谢周期,非法使用会保持其药物原形或代谢产物在畜禽体内残留。消费者食用此产品后会对中枢神经系统造成不良影响,甚至出现增加机体代谢负担和功能性减退等问题,因此许多国家将此类药物列为禁用药物。国际食品法典委员会(CAC)、欧盟等组织和地区均对此类药物推出限量标准[4-5]。我国农业部第235号公告中明确规定氯丙嗪、地西泮等在动物源性食品中不得检出;阿扎哌隆和阿扎哌醇具有相应的参考值;安眠酮禁止使用等[6]。目前镇静剂残留的检测方法主要有酶联免疫法[7]、高效液相色谱法[8-10]、液相色谱-串联质谱法[11-13]和气相色谱-串联质谱法[14-16]。其中酶联免疫法易出现假阳性结果和交叉反应;高效液相色谱法的抗干扰能力差,灵敏度低且不能满足标准限值的要求;GC-MS方法需对样品进行繁琐的衍生化处理;LC-MS/MS技术无需衍生化处理,而且在满足多组分同时检测的情况下仍有较好的选择性、灵敏度和特异性,弥补了前几种方法的缺陷。从实际应用来看,LC-MS/MS方法也是报道最多的方法。

1 实验部分
1.1 仪器与试剂
超高效液相色谱-串联四极杆质谱联用仪(Waters ACQUITY H-CLASS TQD);高速冷冻离心机(美国Thermo公司 MULTIFUGE X1R);涡旋振荡器(美国IKA VORTEX公司 GENIUS 3);超声波清洗器(美国BANDELIN 512H);氮吹仪(Preekem N1公司);天平(感量0.000 1 g,美国Sartorius公司);Milli-Q超纯水器(美国Millipore公司);Oasis MCX 6 cc/150 mg固相萃取柱(美国Waters公司)。
甲醇、乙腈、正己烷(色谱纯,Fisher公司);甲酸(色谱纯,Sigma公司);盐酸、氢氧化钠、氨水(分析纯,北京化工厂);乙酸铵(分析纯,中国国药制剂有限公司);实验用水为超纯水。
1.2 混合标准溶液的制备
分别准确称取上述标准品10.0 mg置于10 mL棕色容量瓶中,加入甲醇溶解并定容至刻度,摇匀,于-20 ℃冰箱保存。临用前,用甲醇稀释成100 ng/mL的混合标准使用液。
1.3 样品前处理
1.3.1 样品预处理 畜肉或肉制品去皮去骨,剔除筋膜,用均质器充分粉碎混匀后,放置-18 ℃以下冷冻贮存。
1.3.2 样品提取 称取2.0 g已粉碎样品(精确至0.01 g)置于50 mL聚丙烯离心管中,加入5 mol/L氢氧化钠溶液0.40 mL,涡旋混合30 s,于65 ℃水浴中放置1 h,期间将样品每隔20 min取出涡旋30 s。水浴后立即加入1.0 mol/L盐酸溶液10.0 mL剧烈振荡20 s,超声10 min,涡旋混合2 min,于-10 ℃,10 000 r/min高速冷冻离心8 min。倾出上清液置于另一50 mL聚丙烯离心管中,在原离心管中再加入1.0 mol/L盐酸溶液7.0 mL重复以上操作,合并上清液并用1.0 mol/L盐酸溶液定容至20 mL。加入10 mL正己烷剧烈振摇2 min,于-10 ℃,10 000 r/min高速离心8 min,吸出下层提取液10.0 mL待净化。1.3.3 净 化 将Waters Oasis MCX固相萃取柱连接固相萃取仪中,移取10 mL提取液过柱,弃去流出液。依次用0.7 mol/L 盐酸溶液3.0 mL、80%甲醇溶液5.0 mL淋洗,用5%氨化甲醇4.0 mL洗脱,收集洗脱液。于40 ℃水浴氮吹至近干,加入流动相定容至1.0 mL,涡旋混合1 min,缓慢过0.22 μm滤膜,滤液待上机测定。
1.4 基质工作曲线配制
依次吸取不同体积的标准使用液加入相同取样量的3种空白基质样品中(牛肉、羊肉、猪肉),样品按“1.3”前处理方法处理后得到3种基质工作曲线,分别对3种样品进行定量校正。16种镇静剂的基质标准工作曲线范围为0.10~10 ng/mL。
1.5 LC-MS/MS测定
1.5.1 色谱条件 Waters BEH C18(2.1 mm×100 mm,1.7 μm)色谱柱;柱温:35 ℃;流速:0.4 mL/min;进样量:5.0 μL;流动相:A为乙腈-0.1%甲酸,B为0.1%甲酸;梯度洗脱程序:0~4.0 min,5%~28% A;4.0~5.5 min,28% A;5.5~8.5 min,28%~30% A;8.5~12 min,30%~68% A;12~16 min,68%~5% A。
1.5.2 质谱条件 电喷雾电离源(ESI+);MRM检测方式;毛细管电压:1.8 kV;脱溶剂气温度:450 ℃;脱溶剂气流量:800 L/h;锥孔气流量:10 L/h;源温度:150 ℃;电离方式、监测离子对、碰撞条件见表1。
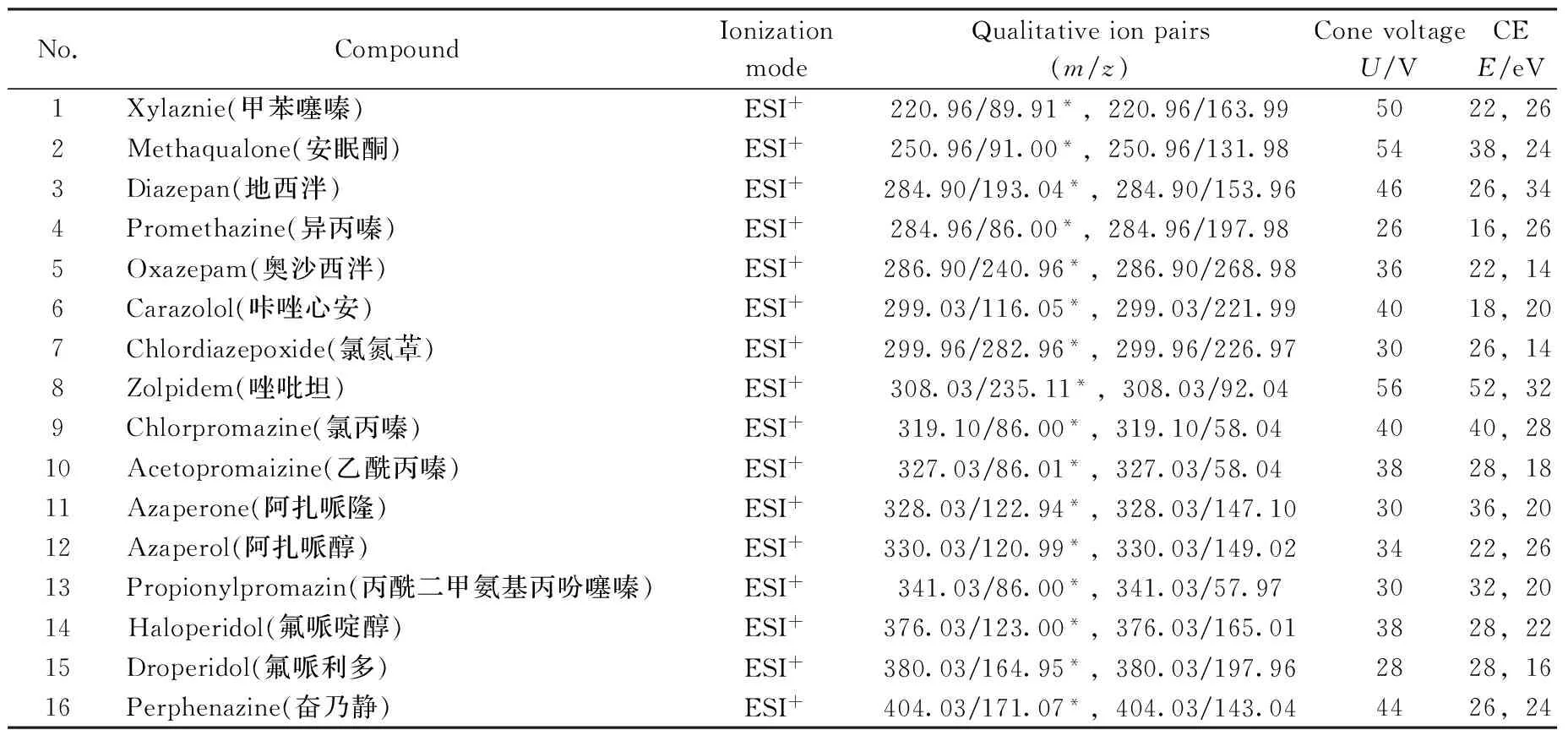
表1 16种待测物质的质谱采集参数Table 1 Mass spectrometric acquisition parameters of 16 compounds
*quantitative ion
1.5.3 样品测定 取基质标准工作曲线分析液和已制备好的样品分析液5 μL分别进样,以峰面积为纵坐标,基质标准工作曲线浓度为横坐标,绘制标准曲线(每个标准点平行测定3次,取峰面积的平均值)。以基质工作曲线标准峰的保留时间和2对离子对作为定性依据,以基质标准工作曲线对样品进行定量。
2 结果与讨论
2.1 色谱条件的优化
本文选择的16种镇静剂多为碱性弱极性化合物[17],对于色谱分离体系中固定相的选择侧重于对弱极性化合物的保留且对流动相pH值有较宽的耐受范围。本方法选择了超高效液相色谱中常用的3种色谱柱(ACQUITY UPLC HSS T3,ACQUITY CSH C18,ACQUITY UPLC BEH C18)进行比较。在使用HSS T3色谱柱时待测物色谱峰有明显的滞后现象,整体时间延长,个别色谱峰展宽明显。分析原因主要是镇静剂类化合物呈碱性,洗脱液加入酸性流动相后弱极性特征发生转变,色谱柱对其保留增强所致。在使用CSH C18色谱柱时色谱峰形尖锐,展宽情况好转且灵敏度提高,但16种镇静剂的色谱峰距离较远,分布不对称。因此,本实验最终选用极性适中的ACQUITY UPLC BEH C18色谱柱,在此条件下各目标化合物的峰形尖锐且基本实现基线分离,具有较好的分离效果和灵敏度。
采用梯度洗脱程序,对超高效液相色谱-质谱系统常用的乙腈-水和甲醇-水流动相体系进行了考察。在使用甲醇-水作为流动相时,柱压明显升高,各组分的保留时间明显滞后。在乙腈-水体系中洗脱性明显增强,各化合物的保留时间缩短,峰形更尖锐且分离度提高。考虑到16种镇静剂的质谱电离方式均为正离子模式,在流动相体系中加入0.1%甲酸溶液后,多个目标化合物的离子强度得到提高,具有较好的洗脱效果和分离度。因此,本实验最终选用0.1%甲酸乙腈-0.1%甲酸水溶液作为流动相,在优化条件下16种镇静剂的总离子流图见图1。
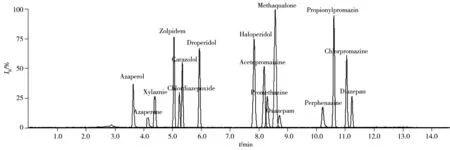
图1 16种化合物混合标准溶液的总离子流图Fig.1 TIC chromatogram of 16 mixed analytes standard solution
2.2 质谱条件的优化
16种待测物的化学结构多为含有氮原子的环状结构,表现出碱性化合物的特性,更适合在质谱ESI源的正电离模式下运行。本实验采用将16种化合物单标液分别进样的方式,在选定的离子化模式基础上,通过全扫描和子离子扫描并调节毛细管电压、锥孔电压、源温度等质谱参数以进一步提高各母离子和子离子的信号水平。根据欧盟2002/657/EC对禁用兽药残留检测确证方法的要求,确证检测需要4个鉴别点。本实验通过上述质谱参数的优化和筛选,每种待测物最终确定2个监测离子对,以满足欧盟兽残检测确证的要求。
2.3 样品前处理条件的优化
为保证样品水解过程的充分性和均匀性,本实验考察了在不同碱液体积,不同水解温度条件下对水解产物最终状态的影响:在80 ℃水浴环境下,分别将0.2,0.4,0.6,0.8,1.0 mL的5 mol/L氢氧化钠溶液加入样品中进行比较。结果显示,加入0.4,0.6,0.8,1.0 mL碱液的样品在50 min内先后全部液化,但加入0.8,1.0mL碱液的样品中出现棕色泡沫,因此确定加入碱液的最佳体积为0.4 mL;同时比较了55,60,65,70,75 ℃水浴温度的影响,结果表明65,70,75 ℃的水浴环境下样品水解完全,液态均匀且颜色透明。因此,最终采用加入5 mol/L氢氧化钠溶液0.4 mL,65 ℃水浴温度作为水解条件。

表2 两种提取方式对基质工作曲线斜率的影响Table 2 Influence of two extractive methods on slopes of the calibration curve with matrix
a:K1is the slope of calibration curve with base;b:K2is the slope of calibration curve with enzyme
2.3.2 净化条件的优化 由于肉类样品基质中脂肪蛋白含量较高,经碱溶液水解后仍残留部分脂肪蛋白干扰。本方法在样品水解液中加入盐酸溶液,使溶液pH值降至蛋白质的等电点以下使蛋白变性后离心沉淀,以达到消除蛋白类的干扰。本实验考察了乙酸乙酯、正己烷、石油醚的去除效果。由于16种待测物中部分呈中等偏弱极性,经实验发现采用乙酸乙酯时待测目标物损失较多,回收率偏低;采用石油醚时,对油脂类和弱极性干扰物的去除效果较好,但样品净化液冷冻离心后,脂肪层出现片状结构且分布不均匀,无法进行萃取操作;而采用正己烷溶剂时,处理液经高速冷冻离心后脂肪层分布均匀且界面清晰,蛋白颗粒全部形成沉淀,待测物损失较少。因此,本方法最终采用正己烷作为净化试剂并结合高速冷冻离心方式,取得了较好的净化效果。由于16种镇静剂多为弱碱性化合物,MCX固相萃取柱对其有较强的吸附作用,因此,本文采用Oasis MCX离子交换固相萃取柱进行净化,通过后续的淋洗和洗脱步骤后,最终获得较为澄清且杂质含量少的样品处理液。
2.4 基质效应的评价及消除
在液相色谱-串联质谱仪的ESI源中,基质效应会影响仪器的灵敏度和重现性。对于肉制品这种富含蛋白、脂肪等复杂基质的样品,其基质效应会更加明显并严重影响检测结果的准确性和重现性。因此在建立本方法的同时对基质效应进行评价,并采取有效措施消除或减弱其影响是非常必要的。本方法采用空白样品处理液加标曲线法(基质匹配标准曲线)评价基质效应(Matrix Effect,ME):ME=(基质匹配标准溶液曲线斜率/无基质标准溶液曲线斜率-1)×100%。基质效应为负值表示存在基质抑制效应;基质效应为正值表示存在基质增强效应;基质效应为0表示无基质效应;绝对值越大表明基质效应越强。本实验在16种镇静剂类待测物曲线范围浓度内进行评价(见表3),可以看出16种镇静剂有不同程度的基质效应,且抑制效应多于增强效应。
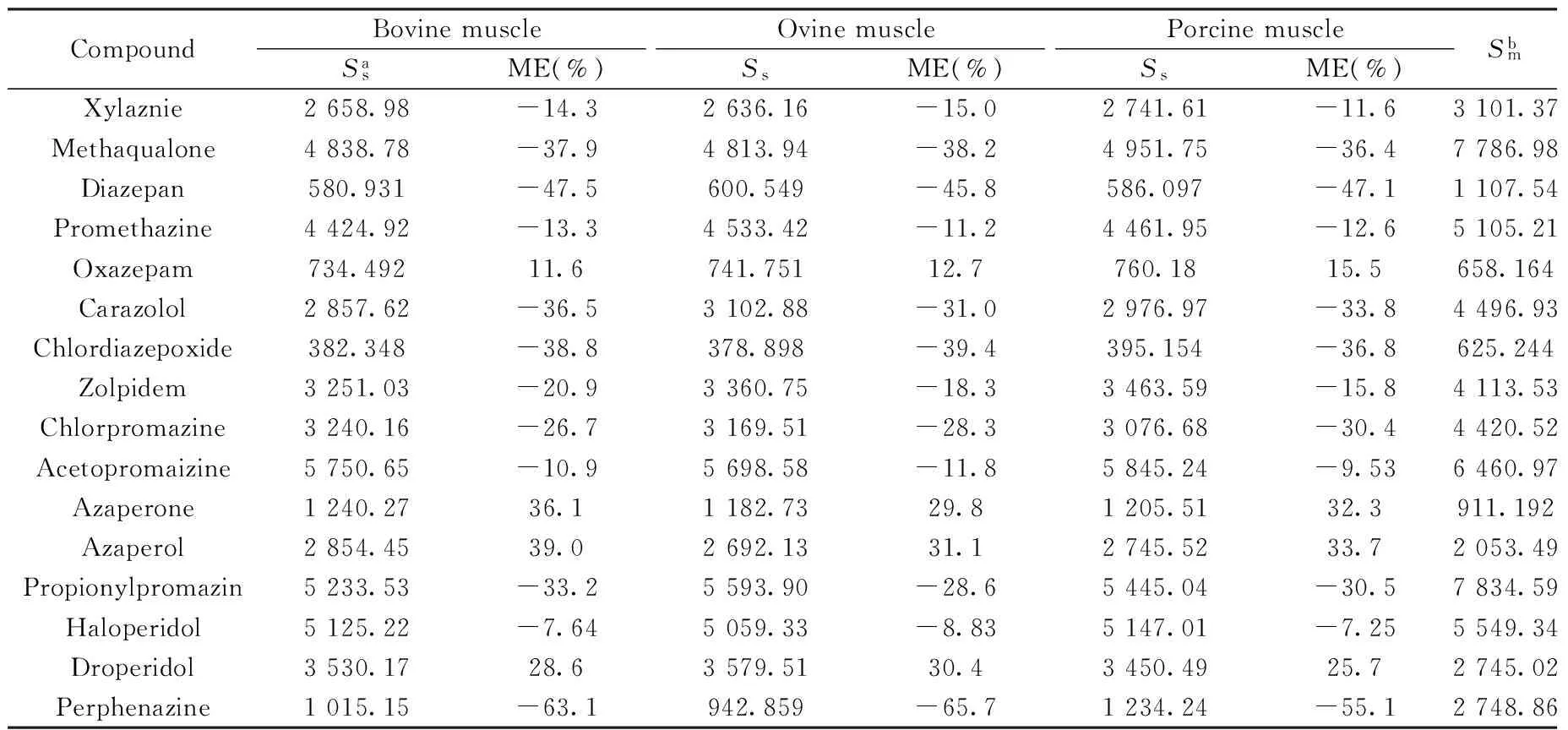
表3 16种待测物质的基质效应Table 3 Matrix effects of 16 compounds
a:Ssis the slope of the calibration curve with matrix;b:Smis the slope of the calibration curve without matrix
为消除基质效应对检测结果的影响,提高检测方法的准确性与重现性,本实验采用基质匹配工作曲线法对各目标物进行定量分析。
2.5 线性范围、检出限与定量下限
本方法采用基质工作曲线作为定量曲线:选取未检出镇静剂的3种阴性畜肉样品(牛肉、羊肉、猪肉),吸取不同量的标准使用液加入3种空白样品中,按照“1.3”步骤处理后经液相色谱-质谱检测,以待测物的峰面积(Y)作为纵坐标,对应的质量浓度(X,μg/L)为横坐标,得到 3种基质的工作曲线。结果显示,16种镇静剂的线性关系良好,相关系数(r2)分别不低于0.996 8。基于基质工作曲线中最低响应的MRM色谱图,确定该方法的检出限LOD(S/N>3)和定量下限LOQ(S/N>10)分别不大于0.05 μg/kg和0.5 μg/kg(结果见表4)。
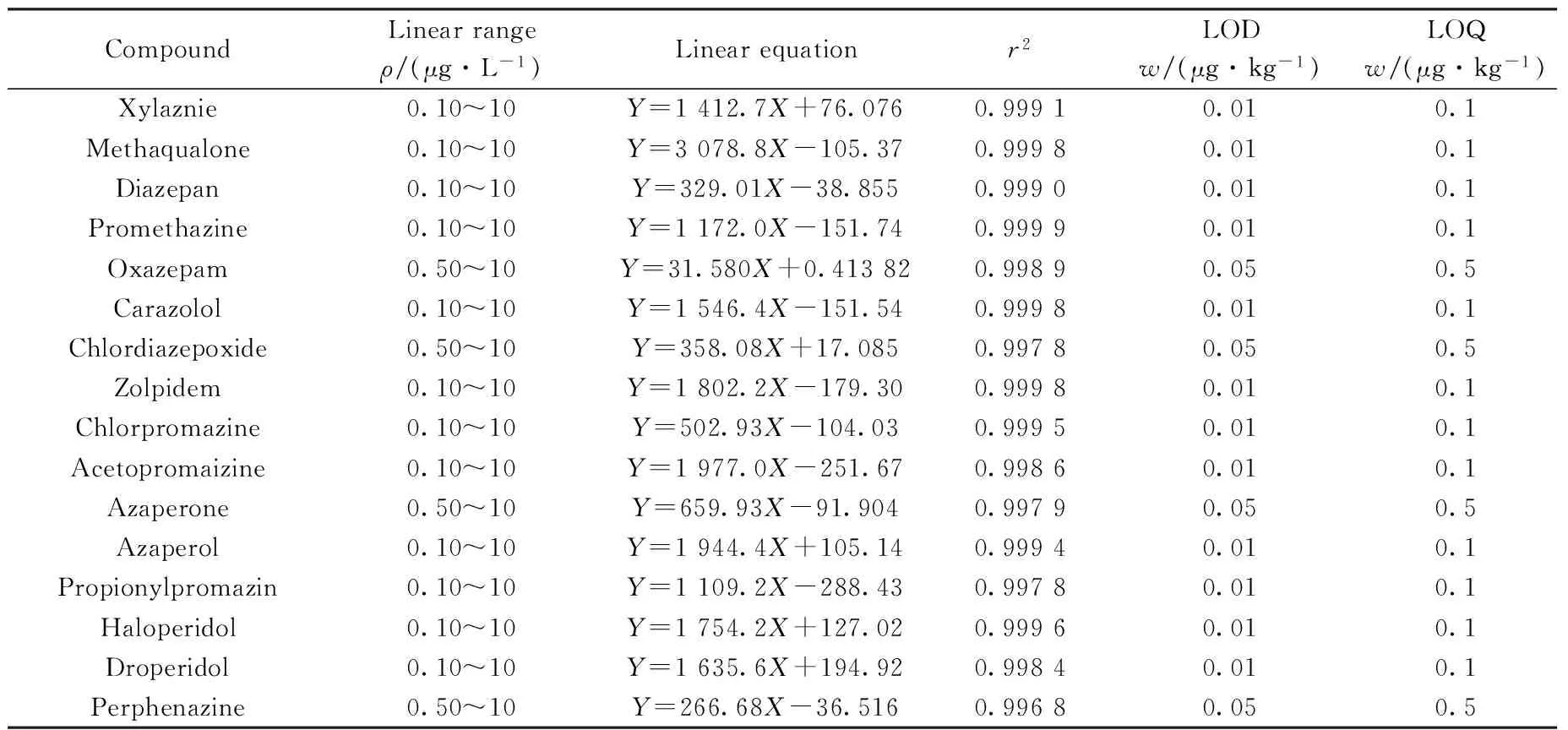
表4 16种镇静剂类化合物的线性范围、线性关系、检出限与定量下限Table 4 Linear ranges,linear equations,LODs and LOQs of 16 compounds
2.6 方法的准确度与精密度
本方法采用加标回收试验进行准确度和精密度的评价。选取不含待测物且充分粉碎混匀的3种畜肉作为空白基质样品,分别按照低(LOQ)、中、高3个浓度水平进行加标回收率测定,每个水平在相同条件下平行测定6个样品,各化合物的回收率和精密度见表5。结果表明,16种镇静剂的平均回收率为71.6%~112%,相对标准偏差(RSD)为2.9%~15.8%。本方法的准确度和精密度均符合有关标准和法规的要求。
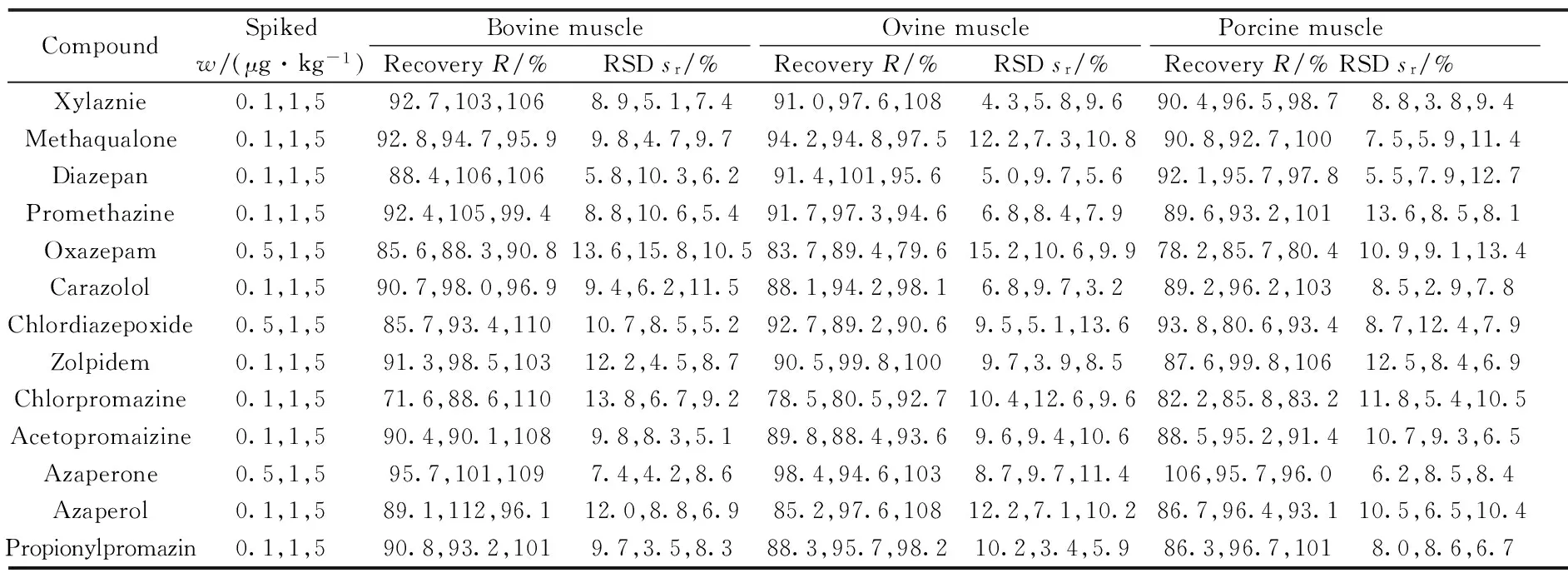
表5 16种镇静剂类化合物的加标回收率及相对标准偏差(n=6)Table 5 Spiked recoveries and RSDs of 16 compounds in blank samples(n=6)
(续表5)

CompoundSpikedw/(μg·kg-1)BovinemuscleOvinemusclePorcinemuscleRecoveryR/%RSDsr/%RecoveryR/%RSDsr/%RecoveryR/%RSDsr/%Haloperidol0.1,1,584.0,102,1106.6,3.8,8.685.2,91.6,1039.7,7.6,8.286.0,94.2,96.19.4,3.4,11.3Droperidol0.1,1,591.2,106,97.76.5,8.8,4.184.2,99.6,10311.5,3.5,8.485.0,102,95.99.9,3.7,8.6Perphenazine0.5,1,597.3,107,1025.6,3.8,8.596.5,106,98.44.8,9.5,4.1102,97.5,1002.9,7.4,5.2
2.7 实际样品的测定
利用本方法对大中型超市、农贸市场、早市的10份畜肉及畜肉制品样品进行检测,均未检出以上16种镇静剂类兽药残留。
3 结 论
本文采用固相萃取的净化方式,建立了超高效液相色谱-串联四极杆质谱测定畜肉及其制品中16种镇静剂类兽药残留的检测方法,该方法采用外标基质工作曲线法定量。在保证样品净化效率的同时有效补偿了基质效应带来的干扰,提高了测定结果的准确性和重现性,通过方法学验证数据和实际抽样检测,证明该方法具有灵敏度高、快速准确、安全经济、适于多组分分析等特点,适用于畜肉及其制品中镇静剂类兽药残留的快速测定。
[1] He J L,Li K X,Yu J L,Liu C J.Chin.AnimalHealthInspect.(贺家亮,李开雄,于见亮,刘成江.中国动物检疫),2006,23(7):22-24.
[2] Yan L J,Zhang J,Pan C S,Lin L Y,Zhang X Y,Shen H Q.Chin.J.Anal.Chem.(严丽娟,张洁,潘晨松,林立毅,张欣怡,申河清.分析化学),2013,41(1):31-35.
[3] Zhang L Q,Wu P G,Jin Q,Zhang Y M,Wang X F,Ren R.Chin.J.HealthLab.Technol.(张力群,吴平谷,金铨,张宜明,王小芳,任韧.中国卫生检验杂志),2013,23(6):1634-1638.
[4] Codex Alimentarius Commission(CAC).Codex Alimentarius Commission Maximum Residue Limits for Veterinary Drugs in Foods Updated as at the 35th Session of the Codex Alimentarius Commission.
[5] European Parliament and Council.Regulation No.470/2009 of the European Parliament and the Council.2009.
[6] Ministry of Agriculture.No.235 Bulletin of the Ministry of Agriculture of the People's Republic of China.(农业部.中华人民共和国农业部公告第235号).
[7] Peng C F,Duan X H,Song S S,Xue F.Int.J.Mol.Sci.,2013,14(10):19474-19483.
[8] Wei J M,Luo Y Z,Bai Y X.Sci.Technol.FoodInd.(魏晋梅,罗玉柱,白云旭.食品工业科技),2014,35(10):95-102.
[9] Li P D,Han H R,Zhai X H,He W H,Sun L Y,Hou J F.J.Chromatogr.Sci.,2012,50(2):108-113.
[10] Liang X H,Dai Z Y,Liu W E.J.CentralSouthUniv.:Med.Sci.(梁湘辉,戴智勇,刘文恩.中南大学学报:医学版),2009,34(7):689-692.
[11] Wei J M,Luo Y Z,Zhang L,Fang S L.J.Sci.FoodAgric.,2015,95(3):598-606.
[12] Sun L,Zhang L,Xu Q,Wang S H,Wang X.Chin.J.Chromatogr.(孙雷,张骊,徐倩,王树槐,汪霞.色谱),2010,28(1):38-42.
[13] Lee S,Han E,In S,Choi H,Chung H,Chung K H.J.Anal.Toxicol.,2011,(35):312-315.
[14] Tan G L,Zhao T Z,Wang W L,Li X L,Zhang N.Mod.FoodSci.Technol.(谭贵良,赵天珍,王文林,李向丽,张娜.现代食品科技),2014,(2):274-278.
[15] Wang L P,Li X,Sun Y,Zhao H X,Qiu Y M,Zhong W K,Tang Y Z,Wang D N,Zhou Z Q.Chin.J.Anal.Chem.(汪丽萍,李翔,孙英,赵海香,邱月明,仲维科,唐英章,王大宁,周志强.分析化学),2005,33(7):951-954.[16] Zhu J,Liu S X,Li B,Han L.J.Instrum.Anal.(朱坚,刘世雄,李波,韩丽.分析测试学报),2005,24(11):301-303.
[17] Zhang S,Zhou S,Chen D W,Hua Z D,Wu Y N,Sun C Y,Zhao Y F.J.Instrum.Anal.(张烁,周爽,陈达炜,花镇东,吴永宁,孙承业,赵云峰.分析测试学报),2014,33(11):1213-1218.
Simultaneous Determination of 16 Sedative Residues in Livestock Products by Solid Phase Extraction/Ultra Performance Liquid Chromatography-Tandem Mass Spectrometry
LIU Jia-yang1,HUANG Xu2,JIA Hong-xin3*
(Liaoning Institute for Food Control,Shenyang 110015,China)
A multi-residue method was established for the simultaneous determination of 16 sedative residues(e.g.xylaznie,methaqualone,diazepan,promethazine,oxazepam,carazolol,chlordiazepoxide,zolpidem,chlorpromazine,acetopromaizine,azaperone,azaperol,propionylpromazin,haloperidol,droperidol,perphenazine) in pork using solid phase extraction/ultra performance liquid chromaography-tandem mass spectrometry(LC-MS/MS).The homogenized samples were hydrolyzed with NaOH,then the analytes were extracted with HCl and cleaned up with an Oasis MCX SPE column.After evaporated to dryness under nitrogen and reconsituted,the reconstituted solution was filtrated through a 0.22 μm syringe filter.Chromatographic separation was performed on a Waters ACQUITY UPLC BEH C18(2.1 mm×100 mm,1.7 μm) column.The qualitative and quantitative detections of 16 analytes were operated by electrospray ionization-tandem mass spectrometry under the positive mode using multiple reaction monitoring(MRM)mode,with the external standard method.The matrix matching external standard method was used for quantitation analysis.All sedative residues have good linear relationship in the certain concentration range with correlation coefficients(r2) not less than 0.996 8.The limits of detection(LOD) and quantitation(LOQ) for 16 sedative residues were in the range of 0.01-0.05 μg/kg and 0.1-0.5 μg/kg,reapectively.The average recoveries at three spiked concentration levels varied from 71.6% to 112%,and the relative standard deviations(RSD) were 2.9%-15.8%.The method was of high sensitivity,simplicity,reliability and low cost,and was suitable for the simultaneous determination of 16 sedative residues in livestock products.
solid phase extraction;ultra performance liquid chromatography-tandem mass spectrometry;sedative;livestock products
10.3969/j.issn.1004-4957.2017.03.002
2016-09-21;
2016-10-12
辽宁省科学事业公益研究基金项目(2014004028)
O657.63;TQ460.72
A
1004-4957(2017)03-0305-07
*通讯作者:贾宏新,博士,主任技师,研究方向:食品安全检验检测,Tel:024-31620803,E-mail:1094888124@qq.com
猜你喜欢
环境保护与循环经济(2021年7期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
食品安全导刊(2021年20期)2021-08-30
中西医结合心血管病电子杂志(2017年6期)2017-09-07
中国实用医药(2016年28期)2016-12-07
当代化工研究(2016年5期)2016-03-20
河北工业科技(2015年4期)2015-02-27
特产研究(2014年4期)2014-04-10
中国现代医生(2014年6期)2014-03-21
食品工业科技(2014年9期)2014-03-11
