
双黄连口服液化学成分含量测定及指纹图谱研究
2017-04-01 06:57冯宏玲李穆睿于思慧
中国民族民间医药 2017年5期
冯宏玲 黄 森 李穆睿 张 慧 * 于思慧 韩 丽
1.辽宁中医药大学,辽宁 大连 116600;2.辽宁省药械审评与监测中心,辽宁 沈阳 110030
双黄连口服液化学成分含量测定及指纹图谱研究
冯宏玲1黄 森2李穆睿1张 慧1*于思慧1韩 丽1
1.辽宁中医药大学,辽宁 大连 116600;2.辽宁省药械审评与监测中心,辽宁 沈阳 110030
目的:测定双黄连口服液化学成分含量,建立化学成分指纹图谱的研究,为双黄连口服液质量标准研究提供理论依据。方法:采用高效液相色谱法,Agilent C18色谱柱(250mm×4.6mm,5μm),流动相为甲醇(A)-0.25%冰乙酸(B),流速为1.0mL/min;柱温为30℃;进样体积为10μL。结果:测定双黄连口服液中化学成分含量并建立了指纹图谱,标定12个共有峰,以9号峰(即黄芩苷)为参照。方法学考察符合规定,RSD均小于3.0%。结论:建立了以黄芩苷、连翘苷和绿原酸为参照物,测定不同厂家的双黄连口服液的含量及化学指纹图谱方法,结果显示不同厂家制剂的指纹图谱存在差异性,此方法简便,重现性好,精密度高,可以为双黄连口服液质量标准提供一定的依据。
双黄连口服液;高效液相色谱法;含量测定;指纹图谱
双黄连口服液是临床抗菌抗病毒常用药,由金银花、黄芩和连翘三味中药提取精制而成,具有抑菌[1]、抗病毒[2-3]、解热抗炎[4]的作用,用于治疗外感风热所致的感冒,症见发热头痛、咳嗽及咽痛等。此三味中药主要成分为绿原酸、黄芩苷以及连翘苷,绿原酸具有抗菌、抗病毒、抗炎、免疫调节等作用[5],黄芩苷具有抗菌抗病毒、解热镇痛抗炎、清除氧自由基等作用[6],连翘苷具有抗菌抗病毒、抗炎、保肝等作用[7],这三种化合物是双黄连口服液中主要活性及药效成分。目前用高效液相色谱法来测定双黄连口服液中的有效成分是主要的检测方式[8-10],且指纹图谱是国内外公认的鉴别中药品种和评价中药质量的非常有效的手段。因此有必要对不同厂家及批次的制剂中这三种成分的含量及指纹图谱进行研究,为此建立了本文的研究方案,可能会对控制药品质量、保证人民用药的安全有效起到积极的作用。
1 仪器与材料
1.1 仪器 Agilent 1200高效液相色谱仪(包括四元泵,DAD 检测器,柱温箱,工作站,安捷伦科技有限公司);AR2140型电子分析天平(上海奥豪斯公司);METTLER AB135-S十万分之一电子天平(瑞士);超声提取仪(240W,50-60Hz,上海必能超声仪器公司)。
1.2 材料 连翘苷(批号:110821-201213)、绿原酸(批号:110753-201314)、黄芩苷(批号:110715-201117)等对照品购自中国食品药品检定研究院。纯净水(杭州娃哈哈集团有限公司),甲醇等试剂均为色谱纯。药品见表1。
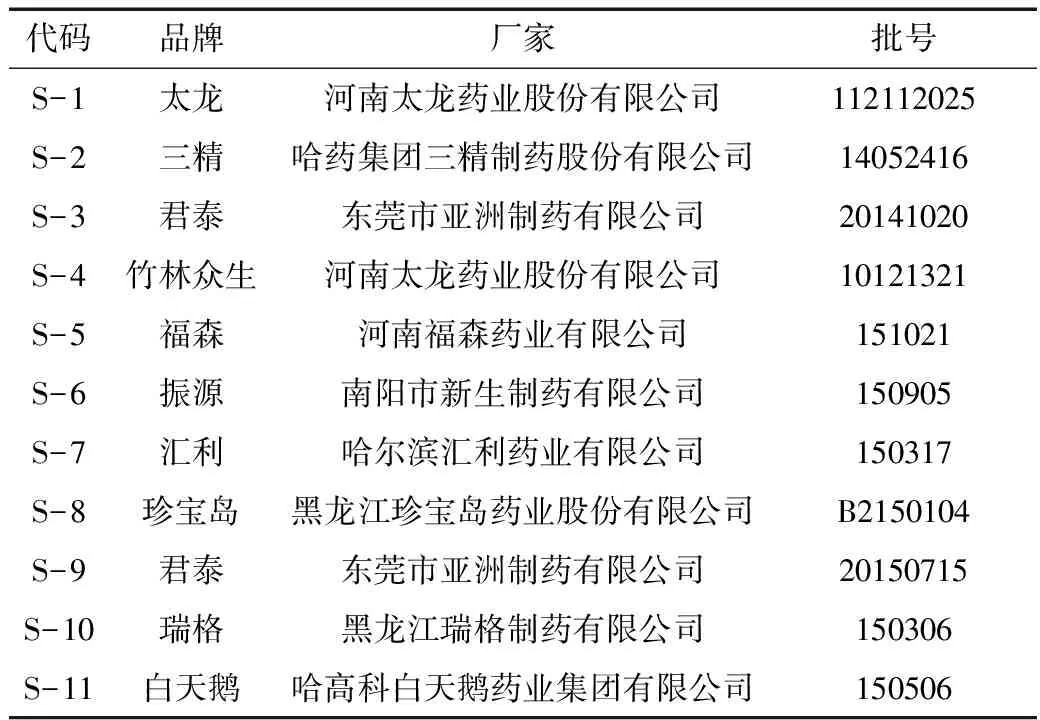
表1 11批双黄连口服液
2 化学成分含量测定
双黄连口服液是由金银花、黄芩、连翘三种药材制成的中药制剂,金银花中有效成分主要为绿原酸、黄芩中有效成分主要为黄芩苷、连翘中有效成分主要为连翘苷,中国药典也以这三种成分作为指标性成分[11],以3种成分为指标,建立了双黄连口服液的含量测定方法。
2.1 色谱条件 梯度洗脱:0~8min,A(15%~30%)-B(85%~70%);8~15min,A(30%~35%)-B(70%~65%);15~20min,A(35%~53%)-B(65%~47%);20~45min,A(53%~63%)-B(47%~37%);45~65min,A(63%~100%)-B(37%~0%);检测波长:280nm。
2.2 对照品溶液制备 分别精密称取绿原酸、连翘苷、黄芩苷对照品,用50%甲醇制成0.1822、0.053、1.18mg/mL的混合对照品溶液,0.45μm微孔滤膜滤过,即得。
2.3 供试品溶液制备 分别取10支双黄连口服液的等量内容物混合,精密量取0.5mL,置于10mL量瓶中,加入50%甲醇定容,摇匀,0.45μm微孔滤膜滤过,即得。
2.4 阴性对照溶液制备 取除去金银花、连翘、黄芩药材,按双黄连口服液制备工艺制备阴性药品,按供试品溶液制备方法操作,制成缺失金银花、连翘、黄芩的阴性对照溶液,备用。
2.5 方法学考察
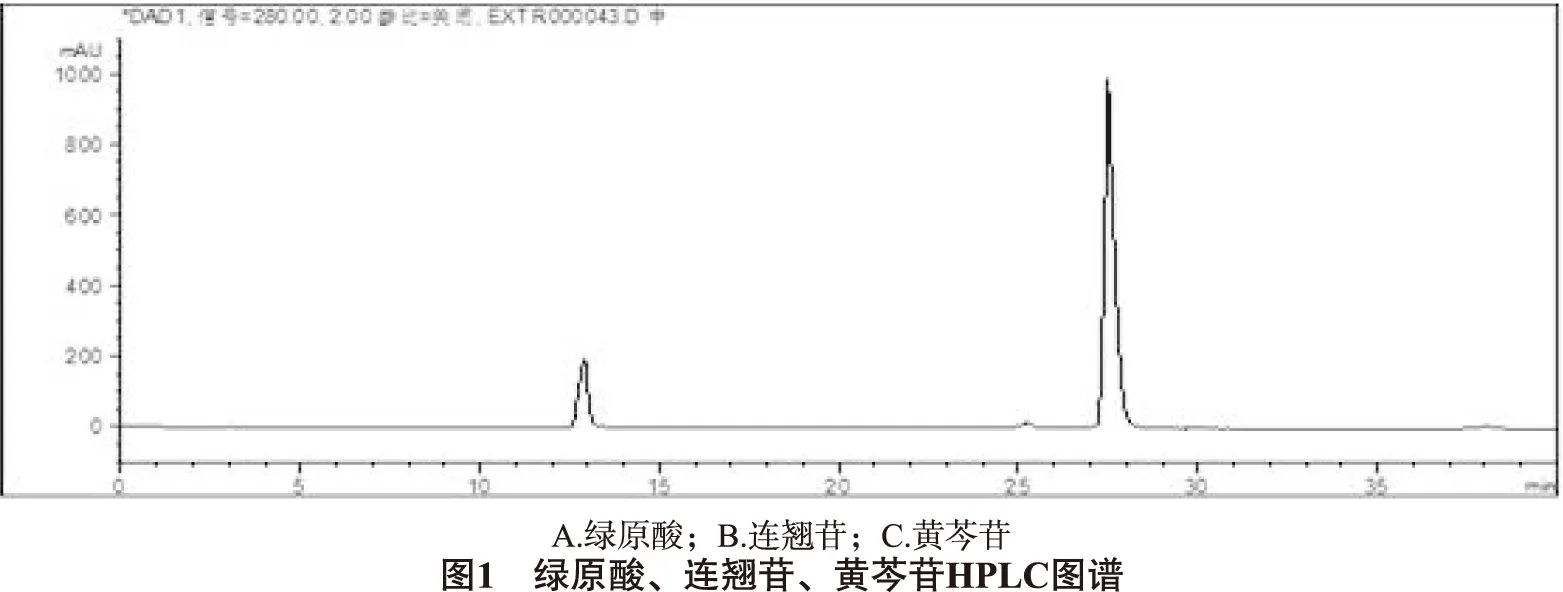
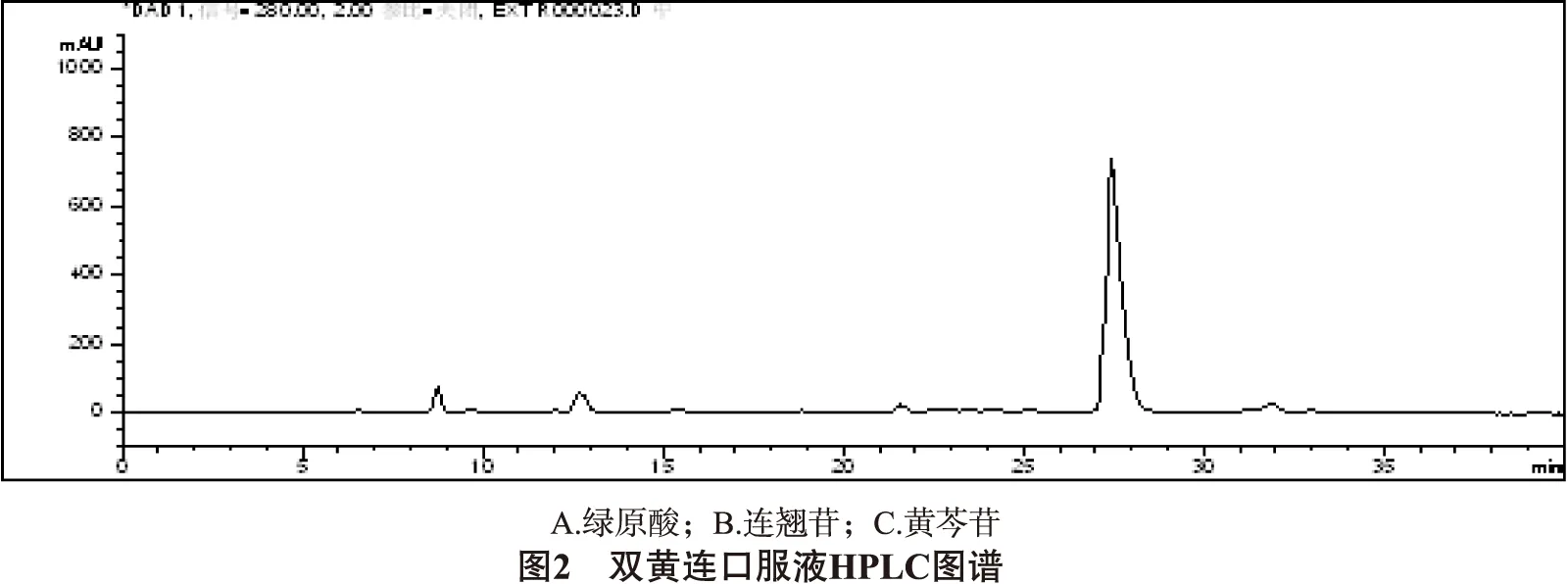
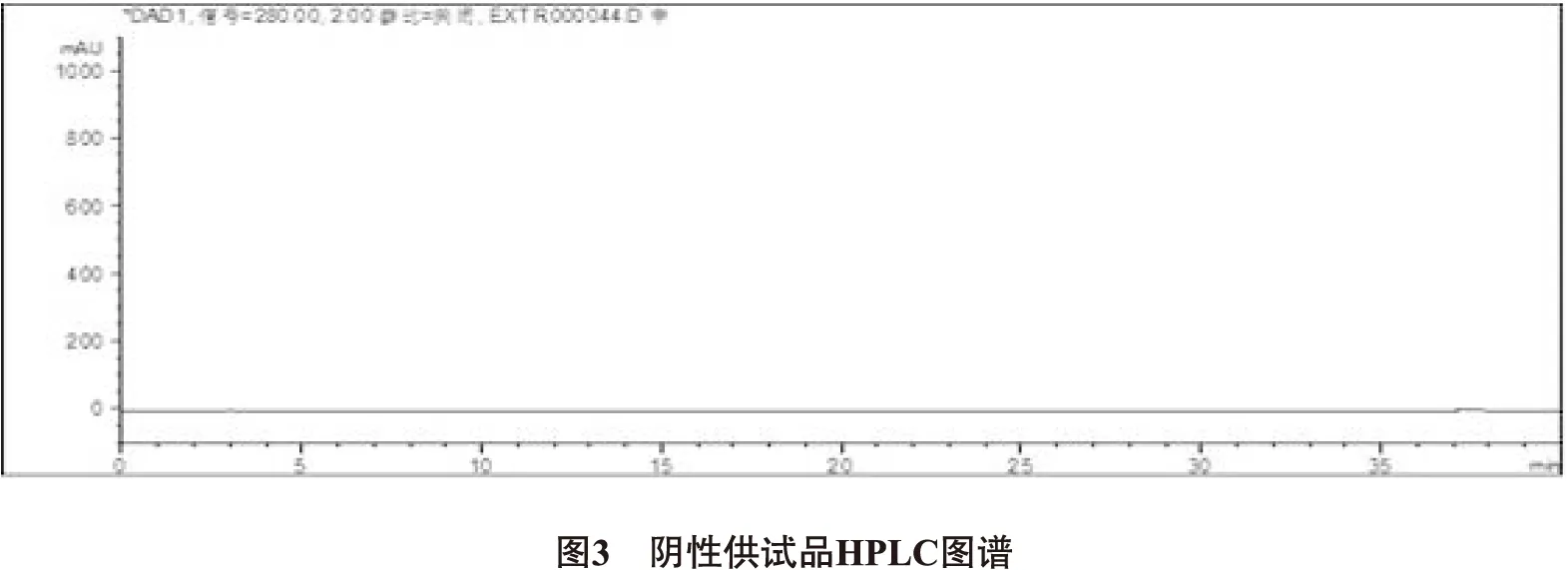
2.5.1 专属性试验 取金银花、连翘、黄芩的阴性对照溶液、对照品溶液、供试品溶液分别注入液相色谱仪,测定色谱峰,结果显示,金银花、连翘、黄芩的阴性溶液色谱中在与供试品色谱中绿原酸、连翘苷、黄芩苷相同保留时间处,未见色谱峰,表明金银花、连翘、黄芩的阴性溶液无干扰。结果见图1、2、3。
2.5.2 线性关系考察 精密量取上述混合对照品溶液2、4、8、10、14、16 μL注入液相色谱仪,测定。以进样量为横坐标,峰面积为纵坐标,绘制标准曲线,计算回归方程。黄芩苷标准曲线方程Y=1890.9X+634.68,R=0.9997,线性范围为2.36μg~18.88μg。精密量取上述混合对照品溶液5、7.5、10、15、20 μL注入液相色谱仪,测定。以进样量为横坐标,峰面积为纵坐标,绘制标准曲线,计算回归方程。绿原酸标准曲线方程Y=128.98X+19.970,R=0.9998,线性范围为0.911μg~3.644μg。连翘苷标准曲线方程Y=12.152X+52.111,R=0.9997,线性范围为0.53~1.06μg。
2.5.3 中间精密度试验 由3个不同实验人员,分别在3天,取双黄连口服液(S-2),精密量取5mL,按“2.3” 项下制备供试品溶液,按“2.1”项下色谱条件,每人重复测定2次,计算含量。结果显示,RSD小于1.5%,表明中间精密度良好。
2.5.4 稳定性试验 取双黄连口服液(S-2),按“2.3” 项下制备供试品溶液,按“2.1”项下色谱条件分别在提取后 0、2、4、6、8、10h取样,分别注入色谱仪,测定色谱峰面积。结果表明,供试品溶液在配制完成后的10h内色谱峰面积无明显变化,表明在10h内供试液溶液稳定。
2.5.5 重复性试验 取双黄连口服液(S-2)6份,按“2.3” 项下制备供试品溶液,按“2.1”项下色谱条件进样分析,测定色谱峰面积,计算含量,RSD小于3%,结果显示,6次测定结果的重复性良好。
2.5.6 准确度试验 精密量取6份已知含量的双黄连口服液(S-2)0.25mL,置10mL量瓶中,分别精密加入黄芩苷(1.18mg/mL)5mL、绿原酸(0.1822mg/mL)5mL和连翘苷(0.053mg/mL) 5ml对照品溶液,按供试品溶液的制备方法制备,注入色谱仪,测定峰面积,计算回收率。结果显示,3个成分的含量的回收率均在95%~105%之间。
2.7 含量测定 按“2.4”项下方法制备供试品溶液,在上述色谱条件下进行测定,采用外标法计算含量。11个不同厂家双黄连口服液含量测定结果有一定差异,其黄芩苷含量在15.055~34.614mg/mL范围内,连翘苷在0.589~2.072mg/mL范围内,绿原酸在0.694~5.781mg/mL范围内,均满足《中国药典》2015版一部的含量限度要求,其中S-7(汇利)3种成分的总量最低。结果见表2。
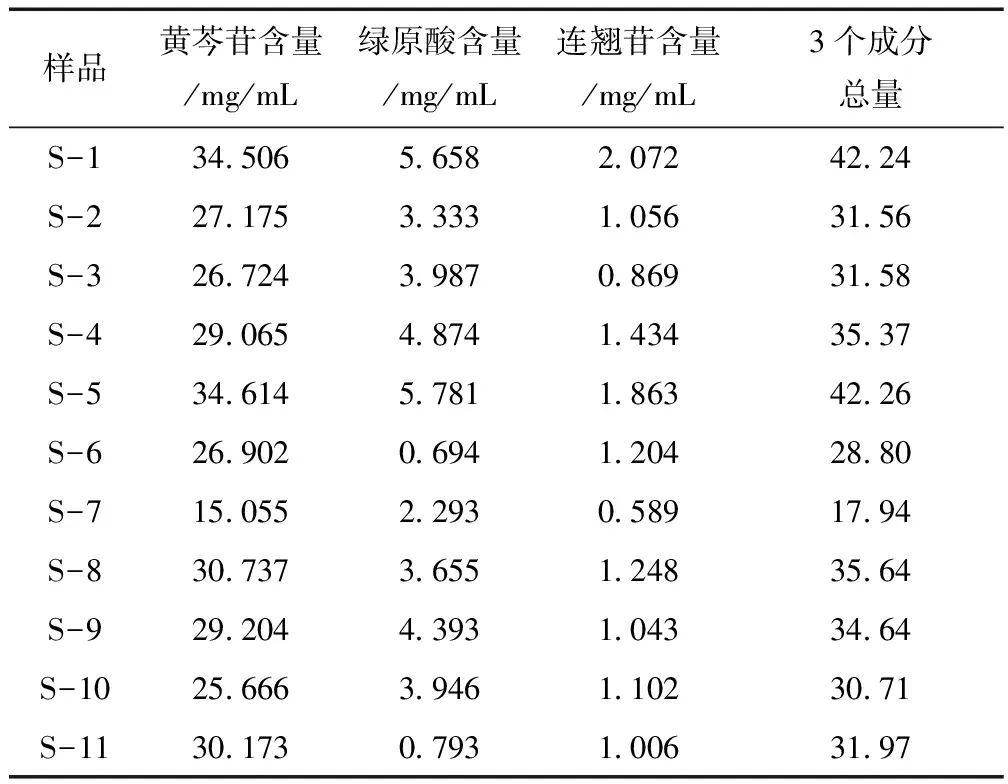
表2 不同厂家批次双黄连口服液中黄芩苷、连翘苷、绿原酸含量测定结果
3 双黄连口服液指纹图谱的建立
3.1 供试品溶液制备 分别取10支双黄连口服液的等量内容物混合,精密量取0.5mL,置于10mL量瓶中,加入50%甲醇定容,摇匀,0.45μm微孔滤膜滤过,即得。
3.2 参照物溶液制备 分别精密称取黄芩苷14.01mg、绿原酸4.24mg、连翘苷5.03mg,置于10mL量瓶中,加入50%甲醇定容,摇匀,分别制得黄芩苷标准品溶液1.401mg/mL、绿原酸0.424μg/mL、连翘苷0.503mg/mL。
3.3 阴性溶液的制备 取除去双黄连口服液,按供试品溶液制备方法操作,制备阴性对照溶液,备用。
3.4 色谱条件 色谱柱:Agilent C18色谱柱(250mm×4.6mm,5μm);流动相:甲醇(A)-0.25%冰乙酸(B);梯度洗脱:0~8min,A(15%~30%)-B(85%~70%);8~15min,A(30%~33%)-B(70 %~67 %);15~20min,A(33%~40%)-B(67%~60%);20~40min,A(40%~43%)-B(60%~57%);40~50min,A(43%~47%)-B(57%~53%);50~70min,A(47%~100%)-B(53%~0%)。流速:1.0mL/min;柱温:30℃;进样体积: 10μL;检测波长:278nm。
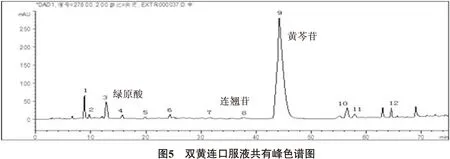
3.5 参照物的确定 通过测定11批不同厂家的双黄连口服液供试品,制剂的色谱峰均在70min之内出现,由于对照品黄芩苷、绿原酸、连翘苷为已知成分,且11批制剂中均存在,因此选择这3个峰作为双黄连口服液的参照峰。参照色谱图见图4。

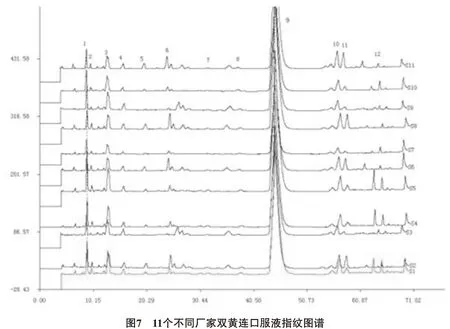
3.6 共有指纹峰的标定 分别测定阴性溶液与供试品溶液,并且进行比较,排除阴性干扰峰,以黄芩苷、绿原酸、连翘苷为参照物,选择11个厂家双黄连口服液指纹图谱中保留时间与参照物相对比的相对保留时间一致的峰做为共有指纹峰,在《中药色谱指纹图谱相似度评价系统》中导入11批不同厂家双黄连口服液的指纹图谱,对保留时间进行相似度评价。结果显示,最终确定共有指纹峰12个,相似度均大于0.99,经计算,每个厂家供试品溶液中色谱图中的共有峰的峰面积之和大于90%,符合共有峰标定要求,共有峰色谱图见图5、6、7,相似度评价见表3。结果显示不同厂家制剂的指纹图谱存在差异性。
3.7 方法学考察
3.7.1 精密度试验 取供试品溶液,连续进样6次,考察共有峰相对保留时间和相对峰面积的值。结果表明,各共有峰的相对保留时间和相对峰面积RSD均小于3%,符合指纹图谱分析要求。在中药色谱指纹图谱相似度评价系统中导入色谱图,得到6个图谱的相似度。结果表明,图谱的相似度均大于0.96,符合指纹图谱分析要求,表明精密度良好。
3.7.2 稳定性试验 取双黄连口服液,按供试品溶液制备方法制备,分别在0、2、4、6、8、10h进样,考察共有峰相对保留时间和相对峰面积的值。结果,各个共有峰相对保留时间和相对峰面积的RSD均小于3%,符合指纹图谱分析要求。在中药色谱指纹图谱相似度评价系统中导入色谱图,得到图谱相似度。结果表明,10h内图谱相似度均大于0.96,符合指纹图谱分析要求,表明稳定性良好。
3.7.3 重复性实验 取同一批号双黄连口服液6份,按供试品溶液制备方法处理,分别注入色谱仪,考察共有峰相对保留时间和相对峰面积的值。结果,各共有峰相对保留时间和相对峰面积RSD均小于3%,符合指纹图谱分析要求。在中药色谱指纹图谱相似度评价系统中导入色谱图,得到图谱相似度。结果表明,图谱的相似度均大于0.97,符合指纹图谱分析要求,表明重复性良好。
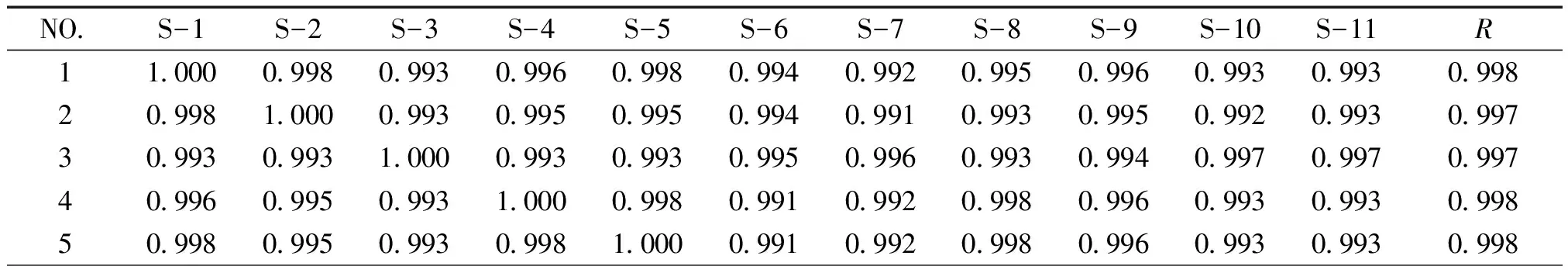
表3 双黄连制剂指纹图谱共有峰相似度评价
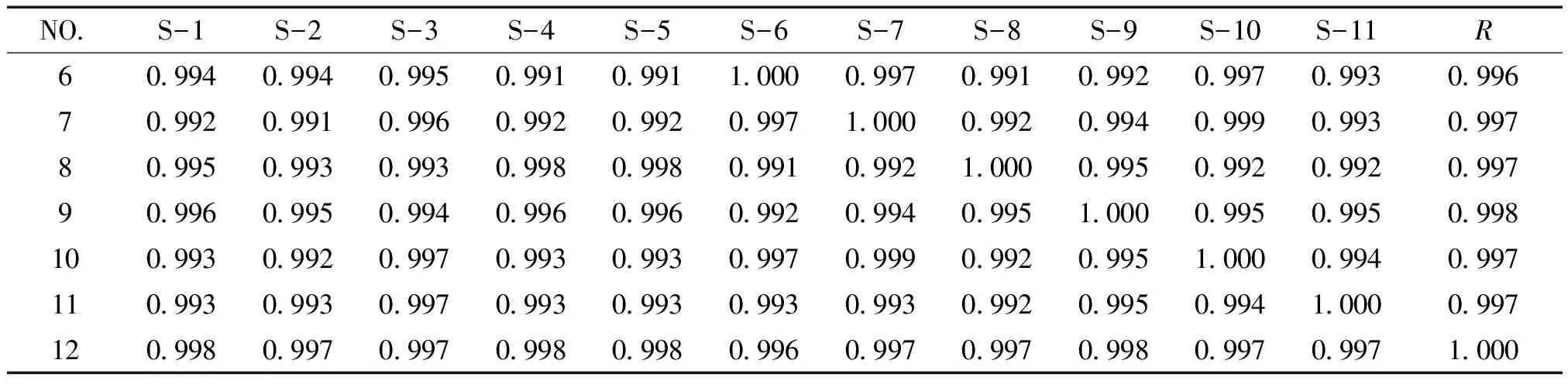
续表3 表3 双黄连制剂指纹图谱共有峰相似度评价
4 讨论
4.1 分别以乙腈-水、甲醇-水等不同比例的流动相系统进行等度和梯度洗脱试验。结果表明,用甲醇-0.25%冰乙酸水溶液进行梯度洗脱可同时将三个有效成分黄芩苷、连翘苷、绿原酸在同一个色谱条件下分开,且分离度较好,为此选用文中流动相。
4.2 本法对双黄连口服液中3种成分同时测定,解决了《中国药典》中采用3个色谱条件分别测定黄芩苷、连翘苷、绿原酸含量的方法的繁琐性,操作简便,提高了双黄连口服液含量测定效率。
4.3 建立了双黄连口服液指纹图谱,标定了12个共有峰,以9号峰(即黄芩苷)为参照,共有峰的相对保留时间分别为:1号(0.200),2号(0.220),3号(0.287),4号(0.358),5号(0.447),6号(0.554),7号(0.713),8号(0.859),9号(1.000),10号(1.285),11号(1.321),12号(1.465),方法学考察符合规定,RSD均小于3.0%;以3号峰(即绿原酸)为参照,共有峰的相对保留时间分别为:1号(0.692),2号(0.760),3号(1),4号(1.233),5号(1.558),6号(1.910),7号(2.481),8号(2.962),9号(3.463),10号(4.432),11号(4.536),12号(5.058),方法学考察符合规定,RSD均小于3.0%。以8号峰(即连翘苷)为参照,共有峰的相对保留时间分别为:1号(0.234),2号(0.257),3号(0.338),4号(0.416),5号(0.526),6号(0.645),7号(0.837),8号(1),9号(1.169),10号(1.496),11号(1.531),12号(1.707),方法学考察符合规定,RSD均小于3.0%。证明此方法可行。
4.4 从11批不同厂家双黄连口服液指纹图谱相似度结果可以看出,不同厂家的双黄连口服液特征图谱相似度较高。但是,各共有特征峰相对峰面积存在差异,说明不同厂家双黄连口服液之间存在质量差异。
[1]徐海瑛,王树芳,王丽, 等.双黄连口服液联合庆大霉素的体外抗菌作用研究 [J].医药导报,2013,32 (1):19-22.
[2] 周雪梦,陆春妮,亓文宝,等.清开灵和双黄连口服液体内抗禽流感病毒作用 [J].中草药,2011,42(7):1351-1355.
[3] 吴成林,杨占秋,侯炜,等.双黄连口服液抗呼吸道合胞病毒的实验研究[J].数理医药学,2005,18(6):592-594.
[4] 邹忠杰,龚梦鹃,王淑美,等.双黄连口服液抗炎作用的代谢组学研究 [J].中成药,2013,35(1):19.
[5] 刘颖,郭明晔,白根本.绿原酸的研究进展[J].中药材,2012,37(7):1182-1183.
[6] 辛文妤,宋俊科,何国荣,等.黄芩素和黄芩苷的药理作用及机制研究进展[J].中国新药,2013,22(6): 647-652.
[7] 魏晋宝,杨光义,陈欢,等.连翘苷的提取方法、药理毒理及药动学研究进展 [J].中国药师,2015,18(12): 2146.
[8] 徐大志,张荣,刘启德, 等.HPLC波长转换法同时检测双黄连口服液中4种有效成分的含量[J].中药新药与临床药理, 2012, 23(1):73-76.
[9] 杜英峰, 张兰桐, 靳怡然,等.多波长RP-HPLC法测定双黄连口服液中黄芩苷、绿原酸和连翘苷 [J].中成药,2009,31(9):1368-1371.
[10] 班丽娜,徐远金.HPLC / MS 同时测定双黄连口服液中的4种有效成分 [J].中成药,2012,34(2):265-267.
[11] 国家药典委员会.中华人民共和国药典(一部)[S].北京:中国医药科技出版社,2015:735-736.
投稿注意事项
文题:中文文题一般以不超过25个汉字为宜。
统计学符号:本刊执行GB3358 1982《统计学名词及符号》的有关规定。统计学符号一律采用斜体表示。
表和图:分别按其在文中出现的先后顺序连续编码,并按先见文后见表(图)的原则排列。每幅表(图)均应冠有表(图)题,说明性的文字应置于表(图)下方的注释中,本刊采用三线表(顶线、表头线、底线)。
Determination of the Chemical Composition and Fingerprint of the Shuanghuanglian Oral
FENG Hongling1HUANG Sen1LI Murui1ZHANG Hui1*YU Sihui1HAN Li1
1.LiaoNing University of Traditional Chinese Medicine,Dalian 116600, China;2.LiaoNing Center for Drug and Device Evaluation and Monitoring,Shenyang 110030,China
Objective The content of chemical constituents in Shuanghuanglian Oral was determined and the fingerprints of chemical constituents were established, which provided the theoretical basis for the quality standard of Shuanghuanglian Oral..Methods Using high performance liquid chromatography, Agilent C18column (250mm×4.6mm, 5μm), the mobile phase of methanol (A) - acetic acid 0.25% Glacial acetic acid B, the flow rate was 1.0mL/min; the column temperature was 30 ℃; the volume of sample is 10μL.Results Shuanghuanglian oral fingerprint was established with 12 common peaks, with 9 peaks (i.e., baicalin) as a reference.The methodological study In accordance with the provisions,RSDwas less than 3.0%.Conclusion Established by baicalin, Phillyrin and chlorogenic acid as reference material,to determine content of Shuanghuanglian oral from different manufacturers and tp establish method of chemical fingerprints, results showed that the fingerprints of different manufacturers have different formulations, this method is simple, reproducible and high precision, which can provide some basis for the quality standard of Shuanghuanglian oral.
Shuanghuanglian Oral ; HPLC;Content Determination; Fingerprint
国家科技部中医药行业科研专项(编号:201407002)。
冯宏玲(1989-),女,汉族,在读硕士研究生,从事中药品质评价及创新药物研究。E-mail:1130048489 @qq.com
张慧(1970-),汉族,博士,教授,从事中药品质评价与创新药物研究。E-mail:syyycs@163.com
R284
A
1007-8517(2017)05-0017-06
2016-12-22 编辑:穆丽华)
猜你喜欢
今日农业(2022年16期)2022-09-22
中国药学药品知识仓库(2022年7期)2022-05-10
右江医学(2022年3期)2022-04-17
今日农业(2021年21期)2022-01-12
当代水产(2021年9期)2021-12-02
西北民族大学学报(自然科学版)(2020年4期)2020-12-21
今日农业(2020年19期)2020-12-14
中国现代医药杂志(2020年10期)2020-12-14
今日农业(2020年16期)2020-09-25
作文新天地(小学版)(2020年5期)2020-06-08
