
黄东海奇异指纹蛤Acila mirabilis和指纹蛤Acila divaricata群体遗传多样性及进化研究*
2017-03-31 06:18胡利莎崔宗梅王海艳
海洋与湖沼 2017年6期
胡利莎 张 振 崔宗梅 王海艳①
(1. 中国科学院海洋研究所 青岛 266071; 2. 中国科学院大学 北京 100049)
奇异指纹蛤Acila mirabilis(Adams and Reeve,1850)和指纹蛤A. divaricata(Hinds, 1843)隶属于胡桃蛤科Nuculidae, 指纹蛤属Acila。因为两者的形态相似性较高, 仅通过形态学特征很难将两者区分开。所以很多研究者认为奇异指纹蛤是指纹蛤的同物异名(Habe, 1958, 1977; Knudsen, 1967; Lutaenkoet al,2012)。但也有很多研究认为它们是两个种(Hanley,1860; Bernardet al, 1993; 徐凤山等, 2008; Huber,2010)。Zhang等(2014)通过形态学和COI基因标记对两者的分类地位进行分析, 认为奇异指纹蛤和指纹蛤为两个不同的种, 二者在形态上也有明显差异: 奇异指纹蛤自壳顶到后腹缘的脊更明显, 脊上通常有一组人字形刻纹; 而指纹蛤这一脊不明显, 脊上的刻纹是壳表刻纹的延续。另外, 奇异指纹蛤和指纹蛤的壳大小不同, 指纹蛤更小, 壳长通常不超过 15mm,奇异指纹蛤壳长可达30mm。对于壳大小相近的样品,指纹蛤铰合齿更加粗壮。奇异指纹蛤和指纹蛤的分布也不同, 奇异指纹蛤分布于黄海、日本北部和俄罗斯远东海, 而指纹蛤分布于东海、南海和日本(徐凤山等,2008)。Huber(2010)和 Zhang等(2014)均认为菲律宾的A. divaricata balabacensis应该也是指纹蛤。另外Zhang等(2014)认为日本所报道的指纹蛤以及日本深海的A. schencki archibenthalis应均为奇异指纹蛤。台湾有关文献记录的指纹蛤大小可达 45mm, 也是奇异指纹蛤(Zhanget al, 2014)。
目前, 有关黄海和东海指纹蛤属物种的研究多集中在群落结构组成的分析上(刘瑞玉等, 1963, 1986;Zhanget al, 2012; 彭松耀, 2013; Liu, 2013; 张鹏弛等,2016), 而对于奇异指纹蛤和指纹蛤的进化关系研究较少。为了解冷水种奇异指纹蛤的群体遗传多样性以及其与指纹蛤的遗传分化水平, 我们在黄海和东海对这二种贝类进行集中采样, 利用线粒体COI(mtCOI)基因进行标记, 从分子水平追踪并探讨指纹蛤属这两个常见种的分化历程和不同群体的遗传多样性, 以期为黄、东海生物多样性研究提供分子生物学基础。
1 材料与方法
1.1 材料与样品制备
实验所需样品采自于黄海、东海, 每群体个体数量为 19—20个, 样品采集后立即用 95%酒精固定,保存备用。图1为样品采集站位的分布, 各采集站位的经纬度和采集数量见表1。冷水团的位置一年四季会有变化, 7月份在南黄海形成的冷中心覆盖本研究涉及的站位, 且3500-10相对靠近冷中心内侧。

图1 奇异指纹蛤和指纹蛤采样站位区域分布图Fig.1 Map of sampling site for A. mirabilis and A. divaricate
1.2 DNA的提取、扩增及测序
取闭壳肌肌肉约100mg, 充分剪碎后, 用TIANamp海洋动物 DNA提取试剂盒(北京天根生物有限公司)提取全基因组DNA。
基因扩增所用引物为 LCO1490 (5′-GGTCAAC AAATCATAAAGATATTGG-3′)和 HCO2198 (5′-AAA CTTCAGGGTGACCAAAAAATCA-3′) (Folmeret al,1994), 由生工生物科技有限公司合成。PCR反应体系 包 括 : 2μL 10×PCR buffer, 1.5mmol/L Mg2+,200μmol/L dNTPs, 1U Taq DNA polymerase, 正、反向引物各0.4μmol/L, 1.5μL 模板DNA溶液(10—100ng/μL), 最后用超纯水补充至25μL。PCR扩增反应程序为: 预变性 94°C 5min; 94°C 30s, 48—52°C 1min,72°C 1min, 循环 30—35 次; 总延伸 72°C 10min。
PCR产物经1%—1.5%琼脂糖电泳凝胶纯化, 送生工生物科技有限公司进行双向测序, 测序引物用扩增时的LCO1490和HCO2198。
1.3 数据分析
DNA序列结果由CLUSTAL X软件(Thompsonet al, 1997)采用默认参数比对, 然后经人工校对后用于分析。
对位后将碱基序列输入DnaSP 5软件(Rozaset al,2003)统计单倍型, 并统计单倍型在各采样站位的分布情况。各物种不同群体的多样性指数, 如单倍型数(h)、核苷酸多样性指数(π)、单倍型多样性指数(H)、平均核苷酸差异数(k)等, 通过Arlequin 3.5 (Excoffieret al, 2010)进行计算。
1.3.1系统发生分析使用ModelTest 3.7 (Posadaet al, 2004)基于 Akaike Information Criterion (AIC)选择核苷酸最佳替代模型为 GTR+I+G。获得的最佳替代模型用于构建最大似然(ML)树和贝叶斯(BI)树, 将近缘的胡桃蛤属Nucula中的橄榄胡桃蛤Nucula tenuis作为外群, 用MrBayes3.2 (Ronquistet al, 2003)软件构建BI树, 用PhyML3.0 (Guindonet al, 2003)软件构建 ML树。另外用软件 Hapstar (Teacheret al,2011)构建单倍型网络关系图来探讨单倍型的谱系结构。NJ树在软件Mega 7 (Kumaret al, 2016)中完成,采用Kimura-2 parameter (K-2P) (Kimura, 1980)计算方法, 用步长值法(Bootstrapping method)估算每个分支1000次重复下的支持度。
1.3.2物种分化时间计算由于缺乏化石或地理记录, 目前还没有针对指纹蛤属贝类计算的序列分歧速率, 本研究采用其他贝类(贻贝属及蚶科物种) mtCOI基因的分歧速率(2%—2.4%/百万年)(Marko, 2002; Markoet al, 2010)进行分析, 推算指纹蛤属物种的分化时间(divergent time,t)。并将指纹蛤属贝类的地理分布与地理历史事件相结合探讨其起源及进化历史。GenBank中已有 COI序列的A.insignis(GenBank number: LC144801)、A. castrensis(KC603848, KC429087)和A. minutoides(LC144800)也加入分析。
使用 BEAST v.1.7.5的宽松分子钟方法(relaxed uncorrelated lognormal clock)估计指纹蛤属起源与分化时间。参数设置为Speciation=Yule process, ngen=10,000,000, log parameter every=1000。TreeAnnotator burn-in=100, 校准点(calibration point)设置为正态分布。最后整合出的系统进化树用 FigTree v1.4查看。
1.3.3群体遗传结构及种群历史动态分析种内群体间遗传分化系数(F-statistics,FST)及其显著性(10000次重复)用Arlequin 3.5软件计算。
中性检验 Tajima’sD(Tajima, 1989a)和 Fu’sFs(Fu, 1997)以及核苷酸不配对分布图(Mismatch distribution)均在Arlequin 3.5软件中计算得到。根据中性检验Tajima’sD和Fu’sFs参数可评估种群是否经历过历史上种群扩张事件。如果D和Fs为显著负值就说明种群经历过历史上扩张(Tajima, 1989b)。此外, 核苷酸不配对分布图也能反应出种群是否经历过扩张(Rogerset al, 1992): 单峰说明种群近期经历过种群扩张; 而多峰则说明种群处于平衡阶段。根据公式τ=2μt估算种群扩张时间,τ可在Arlequin 3.5中得到,µ为序列每代核苷酸的突变速率,t为扩张时间。
2 结果
2.1 系统发生及物种分化时间
测序结果经过人工校对, 共获得 79个奇异指纹蛤个体和60个指纹蛤个体的mtCOI序列, 其长度为641bp(如表 1)。
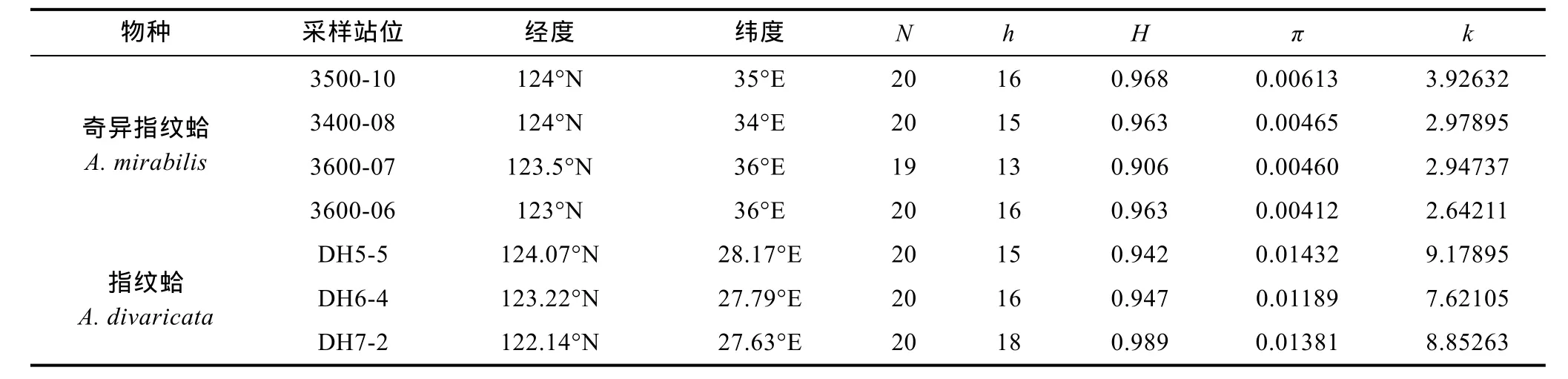
表1 奇异指纹蛤和指纹蛤采样信息及遗传多样性参数Tab.1 The sampling information and genetic parameters of A. mirabilis and A. divaricata
Dnasp分析统计表明, 奇异指纹蛤 79条序列共有50个单倍型, 指纹蛤60条序列共有41个单倍型, 单倍型分布如表2和表3所示。指纹蛤的核苷酸多样性明显高于奇异指纹蛤。奇异指纹蛤群体中3500-10的多样性最高, 3600-06多样性最低。指纹蛤群体中DH5-5的多样性最高, DH6-4多样性最低。
通过PhyML构建的ML树和MrBayes构建的BI树如图2所示, 两种发生树得到的拓扑结构相同, 奇异指纹蛤所有单倍型聚在一起, 指纹蛤所有单倍型聚在一起。
基于奇异指纹蛤所有单倍型和指纹蛤所有单倍型构建的单倍型网络图分别如图3和图4所示, 其结构基本上均呈“星状”。奇异指纹蛤的中间单倍型 Q8的数量最多, 占总数的10.13%, 为群体共有单倍型。指纹蛤的中间单倍型ZA1数目最多, 占总数的20%,为群体共有单倍型。指纹蛤单倍型网络图包括两个“星状”分支, 与单倍型邻接树(图 5)的结果一致, 所以认为指纹蛤单倍型存在A、B两个类群(标记为ZA和 ZB)。
根据 mtCOI 基因 2%—2.4%/百万年的分歧速率计算, 指纹蛤从其余四个种的共同祖先中分化出来的时间大约是 4.27百万年前。奇异指纹蛤从A.insignis、A. castrensis和A. minutoides的共同祖先中分化出来的时间大约是3.71百万年前。A. insignis大约在 1.79百万年前分化出来,A. castrensis和A.minutoides的分化时间大约在1.36百万年前。
指纹蛤两类群 ZA和 ZB的分化时间大约是 64万年前。
2.2 群体遗传结构及种群历史动态
奇异指纹蛤和指纹蛤种内群体间FST结果如表4所示。奇异指纹蛤和指纹蛤群体间均未形成明显的遗传分化, 只有奇异指纹蛤的3500-10与3600-06群体间存在弱的遗传分化。指纹蛤两两群体间的FST存在负值, 说明群体内的遗传差异大于群体间的遗传差异。
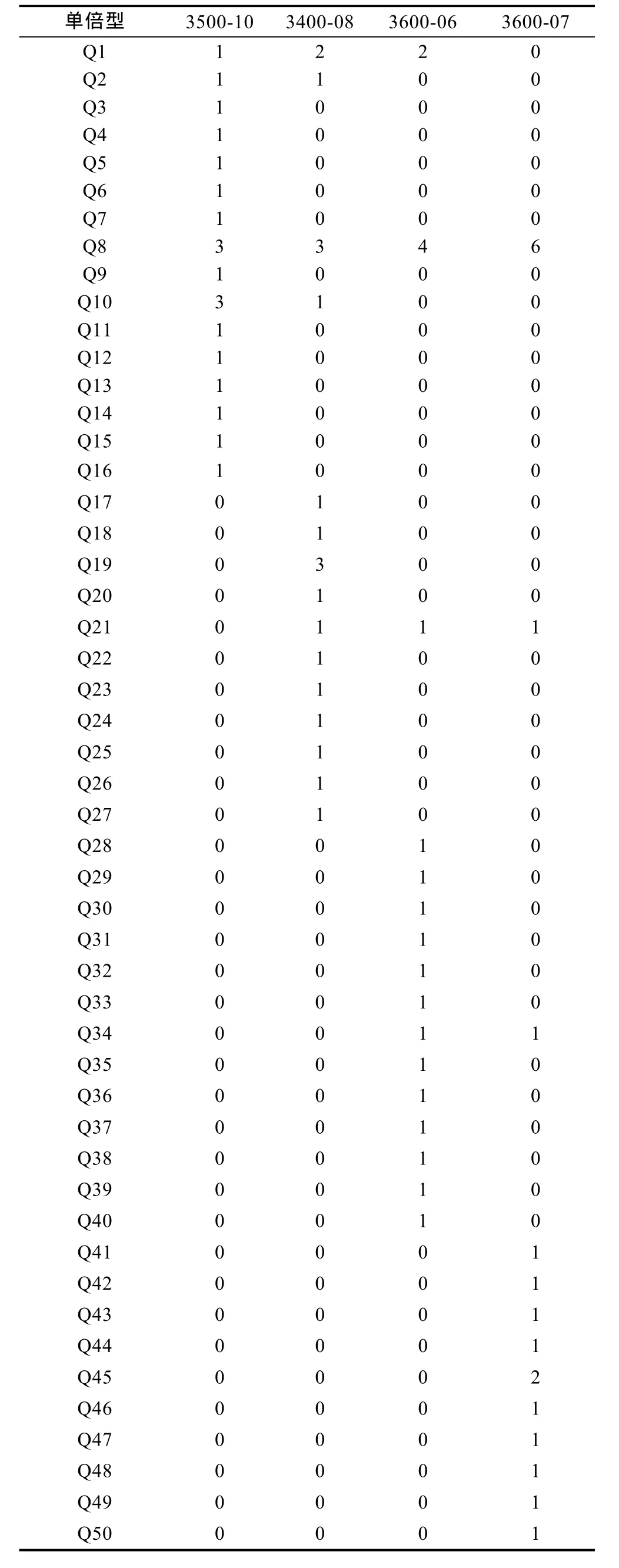
表2 奇异指纹蛤单倍型在各群体中的分布Tab.2 The distribution of haplotypes of A. mirabilis in different populations
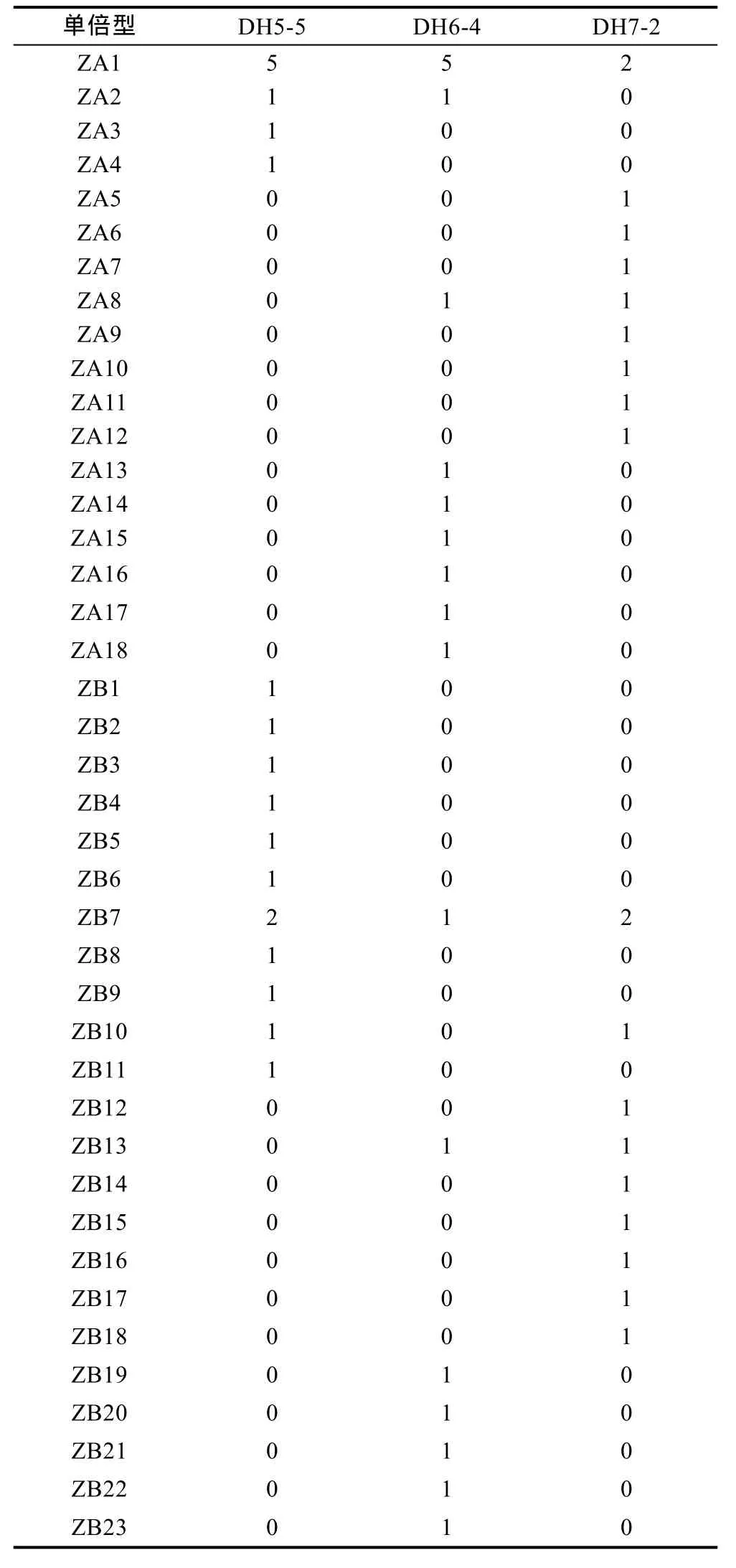
表3 指纹蛤单倍型在各群体中的分布Tab.3 The distribution of haplotypes of A. divaricata in different populations
奇异指纹蛤和指纹蛤各群体的中性检验 Fu’sFs和Tajima’sD结果如表5所示, 奇异指纹蛤各个群体的Fu’sFs及Tajima’sD检验值均为显著负值, 说明在其历史上各群体均经历了种群扩张。
指纹蛤群体只有DH6-4和DH7-2两个群体Fu’sFs为显著负值, 3个群体的Tajima’sD及DH5-5群体的Fu’sFs为负值, 但不显著。
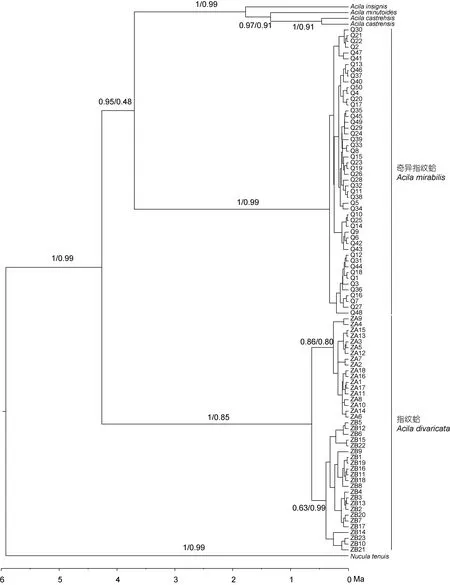
图2 指纹蛤属基于COI单倍型构建的系统发生树Fig.2 Phylogenetic tree of Acila based on COI haplotypes of A. mirabilis and A. divaricata and other sequences downloaded from theGenBank
核苷酸不配对分布如图6和图7所示, 奇异指纹蛤不同群体的核苷酸不配对分布图均为单峰, 也支持种群历史上的扩张。指纹蛤总体及不同群体的核苷酸不配对分布图均为双峰, 其中一个峰对应各类群内序列间的差异, 另一个峰对应两个类群序列间差异。
通过Arlequin 3.5软件计算的τ值(表6所示)可知,奇异指纹蛤3500-10群体最早经历种群扩张, 大约在0.17—0.42百万年前; 3600-06种群扩张时间最晚, 大约在0.09—0.27百万年前。
对指纹蛤 ZA和 ZB两个类群分别统计 Fu’sFs和Tajima’sD, 结果除ZB类群的Tajima’sD不显著外,其余均为显著负值(表 7)。对两个类群分别进行核苷酸不配对分析, 结果均为单峰, 表明两类群均经历了群体扩张(图8)。
根据τ值可知(表 8), 指纹蛤 ZA 类群大约在0.14—0.35百万年前扩张, ZB类群大约在0.33—0.87百万年前扩张。

图3 奇异指纹蛤单倍型网络图Fig.3 The median-joining network of A. mirabilis haplotypes
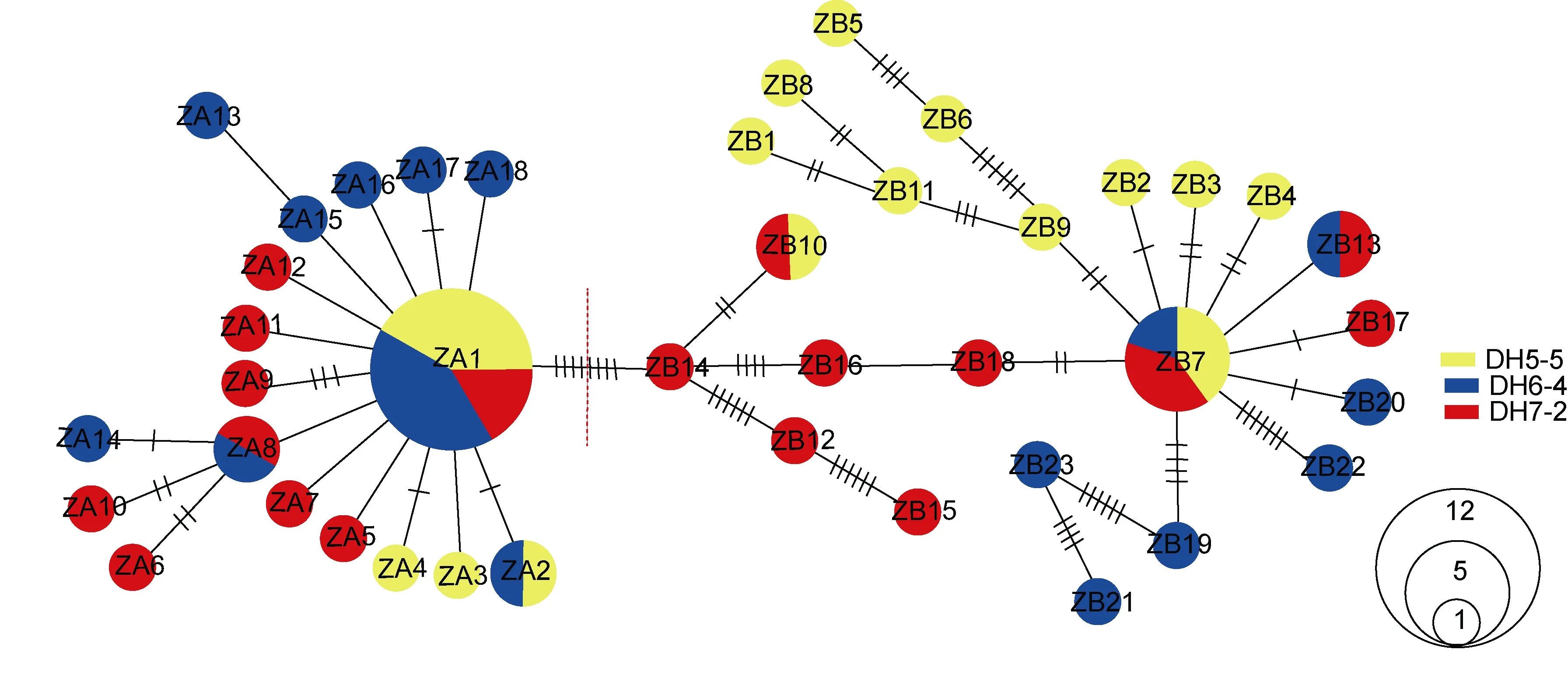
图4 指纹蛤单倍型网络图Fig.4 The median-joining network of A. divaricata haplotypes
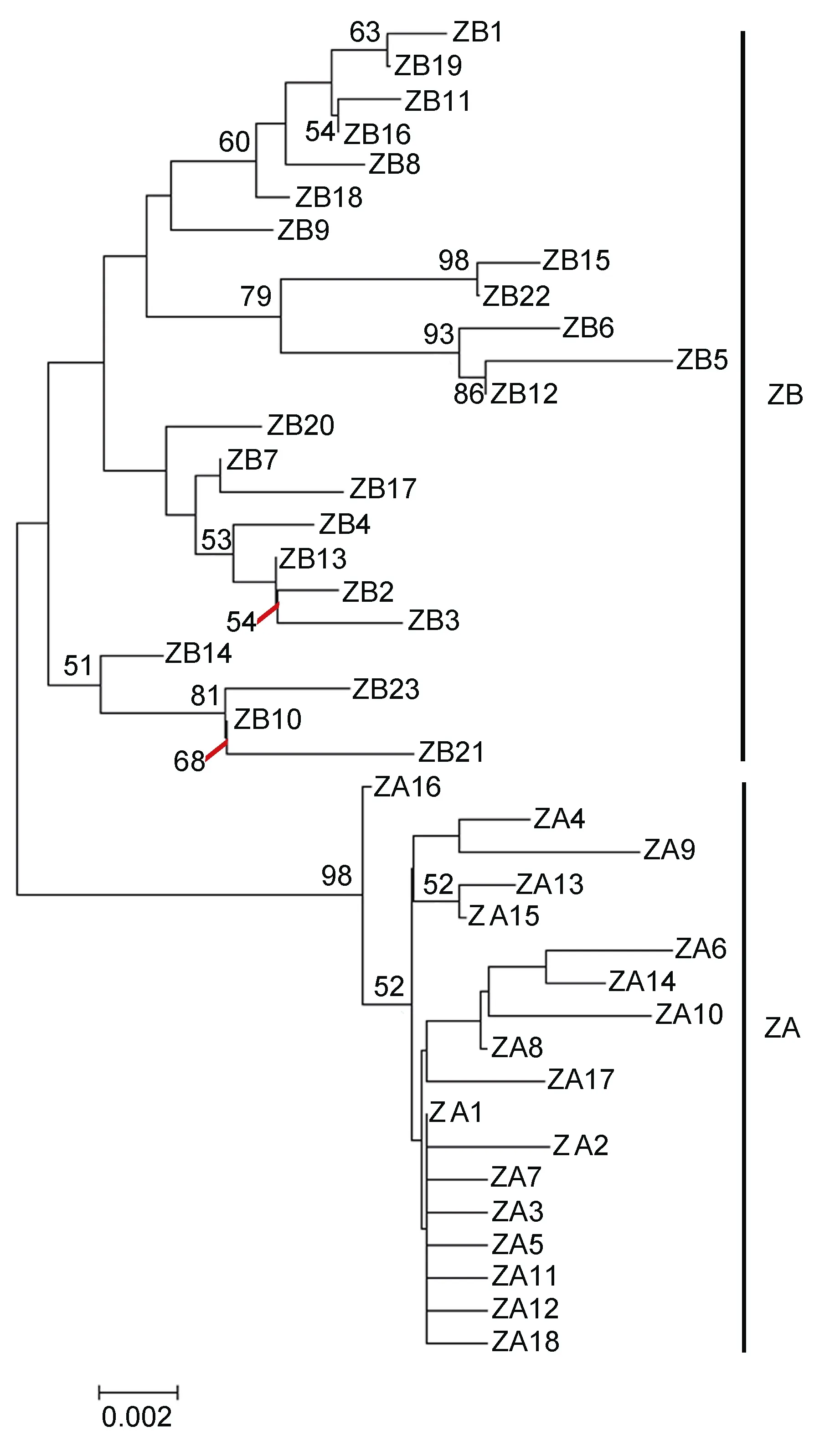
图5 指纹蛤单倍型邻接关系树(NJ树), 支上数值表示bootstrap值>50%Fig.5 The NJ tree of haplotypes of A. divaricata, the numbers at the nodes are bootstrap values >50%
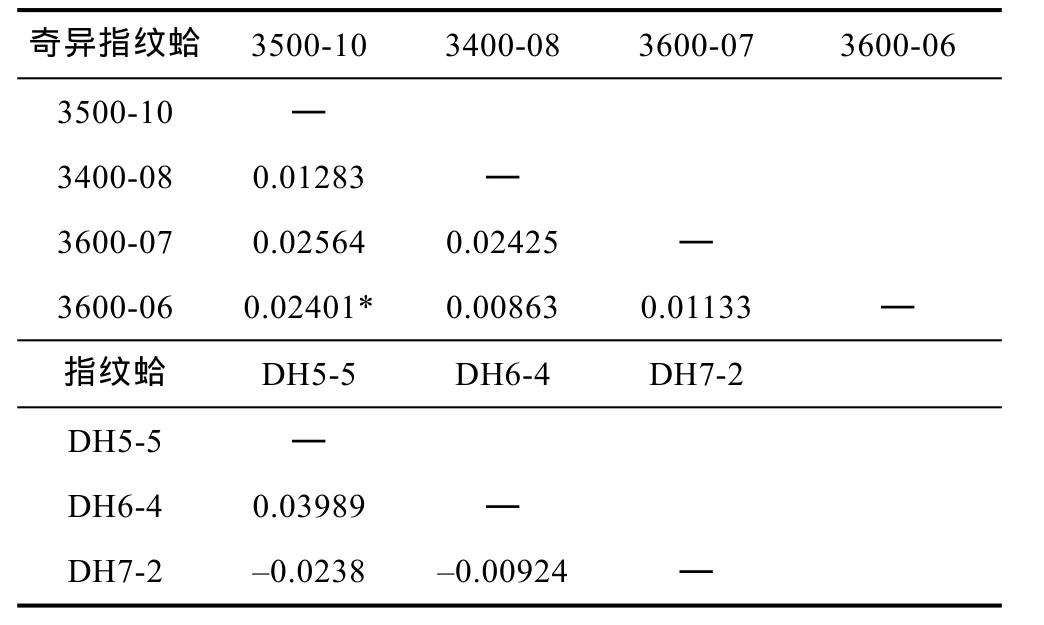
表4 奇异指纹蛤和指纹蛤种内两两群体间FST比较分析Tab.4 FST values between populations within species A.mirabilis and A. divaricata
3 讨论
3.1 指纹蛤属物种分化
线粒体COI基因是动物DNA条形码的有效目标片段, COI序列对于很多种的鉴定来说都是有效的“条形码”(Hebertet al, 2003)。并且其分子进化速率较高, 因而常用于分辨亲缘关系相近的类群(Hebertet al, 2003; Galtieret al, 2009)。本研究以COI基因作为分子标记, 分析结果表明黄海的奇异指纹蛤和东海的指纹蛤为两个不同的种。支持Zhang等(2014)的研究结果。
通过 mtCOI基因的分歧速率计算可知, 在分析的五个种中, 指纹蛤大约在 4.27百万年前最早分化出来, 再依次是奇异指纹蛤(3.71百万年前),A.insignis(1.79百万年前),A. castrensis和A. minutoides分化时间最晚, 大约在1.36百万年前。
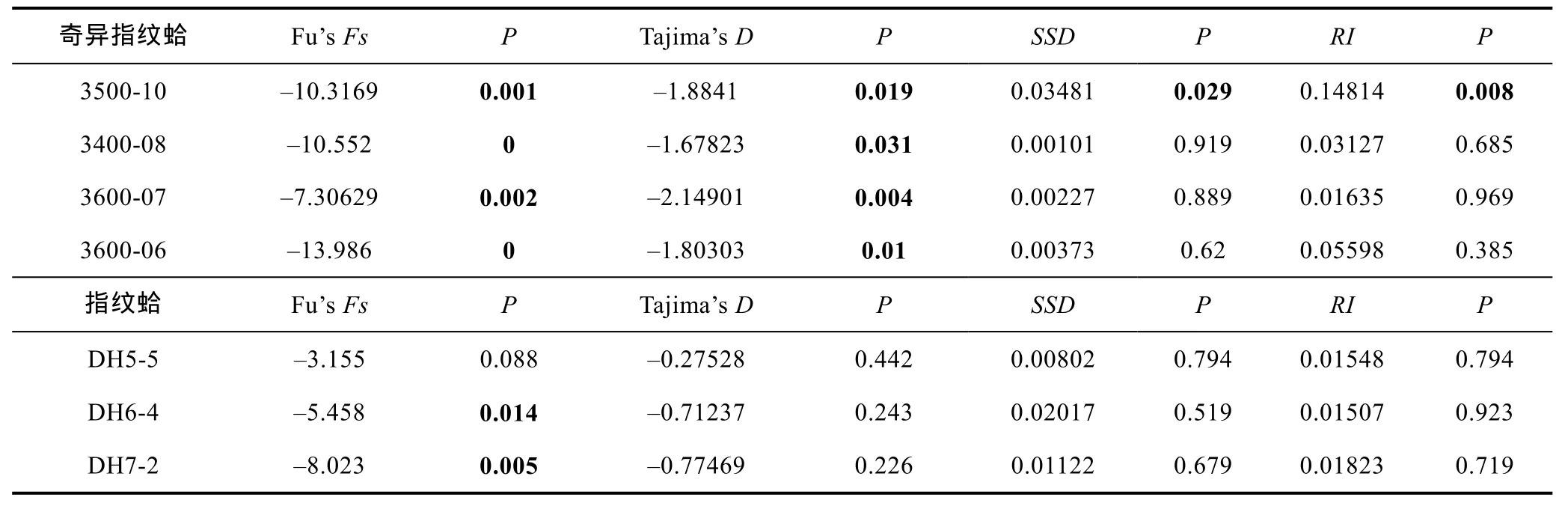
表5 奇异指纹蛤和指纹蛤各群体的Fu’s Fs和Tajima’s DTab.5 Fu’s Fs and Tajima’s D test of populations within species A. mirabilis and A. divaricata
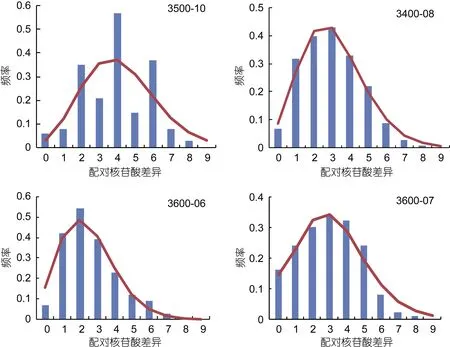
图6 奇异指纹蛤群体核苷酸不配对分布图Fig.6 Pairwise distribution of A. mirabilis populations
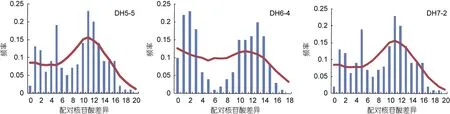
图7 指纹蛤群体核苷酸不配对分布图Fig.7 Pairwise distribution of A. divaricata populations

表6 奇异指纹蛤群体扩张参数Tab.6 Expansion parameters of A. mirabilis populations

表 7 指纹蛤ZA和ZB两类群的Fu’s Fs和Tajima’s DTab.7 Fu’s Fs and Tajima’s D test of ZA and ZB groups within species A. divaricata
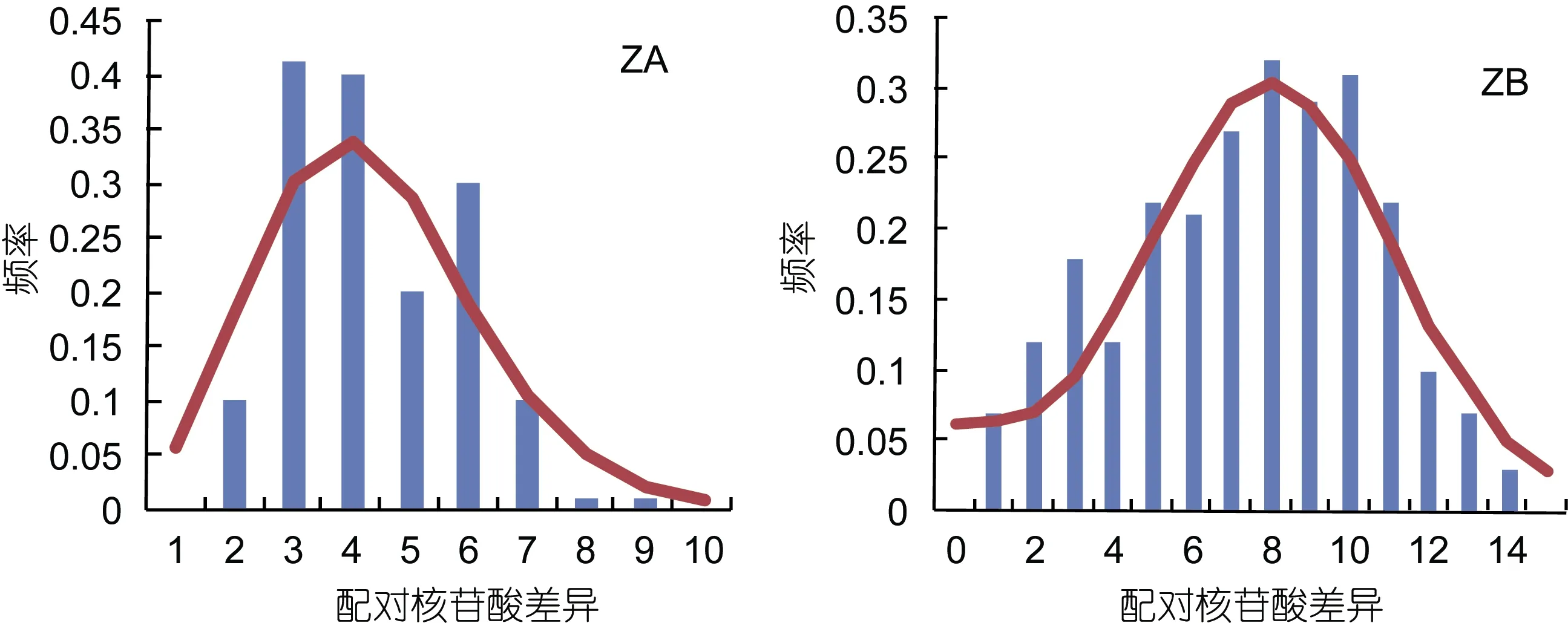
图8 指纹蛤ZA和ZB两类群核苷酸不配对分布图Fig.8 Pairwise distribution of ZA and ZB groups of A. divaricata

表8 指纹蛤ZA和ZB两类群群体扩张参数Tab.8 Expansion parameters of A. divaricata populations
目前很多研究表明, 冰期时幸存在不同海盆或者内海中的海洋生物, 由于相互之间缺乏基因交流,促进了海区间物种的演化, 导致了目前新种或者隐存种的出现。如青蛤分化为三个谱系, 且分别对应三个边缘海(倪刚, 2013)。
指纹蛤和奇异指纹蛤分化出来的时间处于上新世。上新世冰期与间冰期的交替出现显著影响了西北太平洋边缘海的区域和架构(Wang, 1999)。冰期阶段海平面下降, 边缘海之间相互隔离, 不同边缘海内的种群因丧失基因交流而发生分化。我们认为冰期时东海成为指纹蛤属贝类的避难所, 从而使指纹蛤分化形成, 而间冰期海平面上升, 物种向北扩张至黄海,向南扩张至台湾东北部, 扩张至黄海的物种与保留在东海的物种由于环境的差异, 因适应不同的环境而逐渐分化形成两个不同的种, 即本研究中讨论的奇异指纹蛤和指纹蛤, 这一结果与前人的观点(徐凤山等, 2011; Zhanget al, 2014)一致。
目前 WoRMS (http://www.marinespecies.org/)和WMSD (http://www.bagniliggia.it/WMSD/ WMSD home. htm)记录显示A. insignis分布于中国浙江和日本海域, 我们分析的序列来自日本海域的样品。该种的分化时间为 1.79万年前, 这一时间与对马海峡的形成时间(1.52—1.71) (Kitamuraet al, 2001)较接近,这一时期日本海海平面明显低于现在, 洋流仅通过对马海峡涌入日本海, 并无洋流从对马海峡流出(Kitamuraet al, 2006)。因此我们认为对马海峡的屏障作用促进了A. insignis的形成。
A. minutoides的分布水域是中国浙江和日本九州,结合其分化形成时间 1.36百万年前, 推测A.castrensis和A. minutoides的共同祖先由中国迁移至日本, 由于对马海峡的屏障作用, 缺少与中国海之间的基因交流以及对当地环境的适应, 最终导致两种的形成, 其形成也与冰期、间冰期的交替出现有关。
A. castrensis分布于日本、加利福尼亚、加拿大及加伦比亚。我们推测该种可能最先在日本形成, 然后通过海流的作用迁移至目前的这些分布区。这些推测假设需要对该种进行大面积采样, 进一步的分析进而给予较为全面的解释。
综上所述, 就分析的五个种来看, 指纹蛤属的祖先种群可能位于中国东海。后期对指纹蛤属所有现存种进行系统演化及动物地理学研究, 不仅能够验证我们的推测, 对我们理解物种的起源及进化历史也有很大的指导意义。
3.2 目前群体遗传结构的形成
奇异指纹蛤作为黄海常见的冷水种, 其分布也主要集中在冷水团区域及边缘区域。通过分析发现,除 3500-10与 3600-06群体间存在弱的遗传分化外,其余群体间不存在显著的遗传差异。说明其对分布区域适应性强, 群体间存在较强的基因交流。从遗传多样性水平上看, 3500-10群体的多样性水平最高, 且由冷水中心内侧至冷水中心外侧多样性呈递减趋势。除3500-10群体外, 其余3群体的遗传多样性均为高的单倍型多样性(H>0.5)和低的核苷酸多样性(π<0.005), 符合 Grant等(1998)根据不同核苷酸多样性和单倍型多样性间组合的第二种类型, 该类型是由于瓶颈效应后种群扩张以及突变积累所形成, 说明这3个群体经历了群体扩张, 与本研究中单倍型网络图、核苷酸不配对分析以及中性检验结果相一致。而3500-10群体为第四种类型, 这是由于一个大而稳定的种群经过长时间演化所产生或两个不同种群二次接触所造成, 本研究认为是该群体形成后稳定的长时间的演化所产生的。综上所述, 我们推测3500-10可能是本研究中所有奇异指纹蛤类群的祖先群体, 其余群体的形成受冷水团水流的影响(Savolainenet al, 2002)。
指纹蛤主要分布于东海、南海68—162m深水处(徐凤山等, 2008), 因样品数量有限, 本研究仅分析了东海3个群体的遗传多样性及群体遗传结构。研究结果表明, 指纹蛤3个群体的遗传多样性水平均较高,符合 Grant等(1998)提出的第四种类型, 这可能是由于不同种群二次接触造成的, 即由历史上发生过异域分化, 随后分化的类群间再次混合发生二次接触形成(Avise, 2000)。指纹蛤单倍型网络图及中性检验结果均支持这一推测, 即每个群体内都存在2个分化的类群。在一些鱼类和蟹类的研究中也有与该模式类似的报道(Liuet al, 2007; Wanget al, 2008; 殷维,2010)。根据 mtCOI基因2%—2.4%/百万年的分歧速率计算, 指纹蛤两类群ZA和ZB的分化时间大约是64万年前, 发生于更新世中期。冰期时, 海平面发生强烈的变化, 尤其是在冰期盛行时期, 海平面下降最大, 约120—140m (Lambecket al, 2002)。冰期海平面下降, 东海大陆架裸露成为陆地, 海洋生物的生物量减少, 只有少数幸存个体残存于有限的避难所内, 而临时避难所的相互隔离, 最终导致了指纹蛤群体产生分化。冰期过后, 海平面上升, 指纹蛤可能由冰期时的临时避难所发生殖化事件, 从而使得先前被隔离的群体间发生混合, 从而形成了目前的群体结构。另外, 一个大而稳定的群体经过长时间演化也可能出现显著的分化。结合贝类成体前的幼体浮游阶段的生活史, 我们认为这也可能是在指纹蛤群体内检测到两个分化类群的原因。
我们发现指纹蛤的 3个群体位于黑潮分支流-近岸黑潮底层分支(NKBC)流经区域附近, 所以我们认为东海大面积的群体采样以及补充南海群体样品进行指纹蛤详细的群体遗传结构的分析, 将会更好地阐释其进化模式。
4 结论
本研究结果支持前人的研究, 即奇异指纹蛤和指纹蛤是指纹蛤属在黄海和东海不同海域分布的两个不同种。两种的分化可能与冰期时海平面的升降及环境的适应性有关。奇异指纹蛤和指纹蛤不同群体间不存在显著的差异, 而指纹蛤单倍型存在两个分化的类群, 说明其在历史上经历过隔离分化。
刘瑞玉, 徐凤山, 1963. 黄、东海底栖动物区系的特点. 海洋与湖沼, 5(4): 306—321
刘瑞玉, 崔玉珩, 徐凤山等, 1986. 黄海、东海底栖生物的生态特点. 海洋科学集刊, 27: 153—173
张鹏弛, 徐 勇, 李新正等, 2016. 南黄海夏季软体动物群落结构特征. 海洋与湖沼, 47(4): 774—786
倪 刚, 2013. 四种广布性贝类系统地理学研究——探讨中国近海贝类遗传格局形成过程和演化机制. 青岛: 中国海洋大学博士学位论文
徐凤山, 张均龙, 2011. 中国海典型生境双壳类软体动物多样性特点. 生物多样性, 19(6): 716—722
徐凤山, 张素萍, 2008. 中国海产双壳类图志. 北京: 科学出版社
殷 维, 2010. 西北太平洋边缘海厚蟹属的物种界定与历史地理学. 上海: 复旦大学硕士学位论文
彭松耀, 2013. 黄东海大型底栖动物群落结构特征. 青岛: 中国科学院研究生院(海洋研究所)博士学位论文
Avise J C, 2000. Phylogeography: The History and Formation of Species. Oxford: Harvard University Press, 1—361
Bernard F R, Cai Y Y, Morton B, 1993. Catalogue of the Living Marine Bivalve Molluscs of China. Hong Kong: Hong Kong University Press, 146
Excoffier L, Lischer H E L, 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources,10(3): 564—567
Folmer O, Black M, Hoeh Wet al, 1994. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology, 3(5): 294—299
Fu Y X, 1997. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection.Genetics, 147(2): 915—925
Galtier N, Nabholz B, Glémin Set al, 2009. Mitochondrial DNA as a marker of molecular diversity: a reappraisal. Molecular Ecology, 18(22): 4541—4550
Grant W A S, Bowen B W, 1998. Shallow population histories in deep evolutionary lineages of marine fishes: insights from sardines and anchovies and lessons for conservation. Journal of Heredity, 89(5): 415—426
Guindon S, Gascuel O, 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology, 52(5): 696—704
Habe T, 1958. Report on the Mollusca chiefly collected by the S.S. Soyo-Maru of the Imperial Fisheries Experimental Station on the continental shelf bordering Japan during the years 1922—1930. Part 3. Lamellibranchia (1). Publications of the Seto Marine Biological Laboratory, 6(3): 241—280
Habe T, 1977. Systematics of Mollusca in Japan: Bivalvia and Scaphopoda. Tokyo: Hokuryukan Publishing Co., 372
Hanley S C T, 1860. Monograph of the family Nuculidæ forming the Lamarckian genusNucula. In: Sowerby II G B ed.Thesaurus Conchyliorum, or, Monographs of Genera of Shells, Vol.3. London: Palala Press, 105—168
Hebert P D N, Cywinska A, Ball S Let al, 2003. Biological identifications through DNA barcodes. Proceedings of the Royal Society B: Biological Sciences, 270(1512): 313—321
Huber M, 2010. Compendium of Bivalves: A Full-Color Guide to 3300 of the World’s Marine Bivalves: A Status on Bivalvia after 250 Years of Research. Hackenheim: ConchBooks, 901
Kimura M, 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution 16(2):111—120
Kitamura A, Kimoto K, 2006. History of the inflow of the warm Tsushima Current into the Sea of Japan between 3.5 and 0.8 Ma. Palaeogeography, Palaeoclimatology, Palaeoecology,236(3—4): 355—366
Kitamura A, Takano O, Takata Het al, 2001. Late Pliocene-early Pleistocene paleoceanographic evolution of the Sea of Japan.Palaeogeography, Palaeoclimatology, Palaeoecology,172(1—2): 81—98
Knudsen J, 1967. The Deep-sea Bivalvia. The John Murray Expedition, 1933-34, Scientific Reports, Vol.XI, No.3.British Museum (Natural History), 239—343, 1—3pl (in Japanese)
Kumar S, Stecher G, Tamura K, 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets.Molecular Biology and Evolution, 33(7): 1870—1874
Lambeck K, Esat T M, Potter E K, 2002. Links between climate and sea levels for the past three million years. Nature,419(6903): 199—206
Liu J X, Gao T X, Wu S Fet al, 2007. Pleistocene isolation in the Northwestern Pacific marginal seas and limited dispersal in a marine fish,Chelon haematocheilus(Temminck &Schlegel, 1845). Molecular Ecology, 16(2): 275—288
Liu J Y, 2013. Status of marine biodiversity of the China Seas.PLoS One, 8(1): e50719
Lutaenko K A, Noseworthy R G, 2012. Catalogue of the Living Bivalvia of the Continental Coast of the Sea of Japan (East Sea). Vladivostok: Dal’nauka, 247
Marko P B, 2002. Fossil calibration of molecular clocks and the divergence times of geminate species pairs separated by the Isthmus of Panama. Molecular Biology and Evolution,19(11): 2005—2021
Marko P B, Hoffman J M, Emme S Aet al, 2010. The 'Expansion-Contraction’ model of Pleistocene biogeography: rocky shores suffer a sea change? Molecular Ecology, 19(1): 146—169
Posada D, Buckley T R, 2004. Model selection and model averaging in phylogenetics: advantages of akaike information criterion and Bayesian approaches over likelihood ratio tests. Systematic Biology, 53(5): 793—808
Rogers A R, Harpending H, 1992. Population growth makes waves in the distribution of pairwise genetic differences.Molecular biology and evolution, 9(3): 552—569
Ronquist F, Huelsenbeck J P, 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics,19(12): 1572—1574
Rozas J, Sánchez-DelBarrio J C, Messeguer Xet al, 2003. DnaSP,DNA polymorphism analyses by the coalescent and other methods. Bioinformatics, 19(18): 2496—2497
Savolainen P, Zhang Y P, Luo Jet al, 2002. Genetic evidence for an East Asian origin of domestic dogs. Science, 298(5598):1610—1613
Tajima F, 1989a. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics,123(3): 585—595
Tajima F, 1989b. The effect of change in population size on DNA polymorphism. Genetics, 123(3): 597—601
Teacher A G F, Griffiths D J, 2011. HapStar: automated haplotype network layout and visualization. Molecular Ecology Resources, 11(1): 151—153
Thompson J D, Gibson T J, Plewniak Fet al, 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools.Nucleic Acids Research, 25(24): 4876—4882
Wang C H, Li C H, Li S F, 2008. Mitochondrial DNA-inferred population structure and demographic history of the mitten crab (Eriocheir sensu stricto) found along the coast of mainland China. Molecular Ecology, 17(15): 3515—3527
Wang P X, 1999. Response of Western Pacific marginal seas to glacial cycles: paleoceanographic and sedimentological features. Marine Geology, 156(1—4): 5—39
Zhang J L, Shi H F, Xu F Set al, 2014. AreAcila divaricataandAcila mirabilisOne species or two distinct species?Evidence from COI mitochondrial DNA. Journal of Ocean University of China, 13(2): 283—289
Zhang J L, Xu F S, Liu R Y, 2012. Community structure changes of macrobenthos in the South Yellow Sea. Chinese Journal of Oceanology and Limnology, 30(2): 248—255
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
生态学报(2021年3期)2021-03-31
绿色科技(2019年14期)2019-11-19
江苏农业科学(2019年5期)2019-09-02
中华诗词(2019年2期)2019-08-27
中国饲料(2019年19期)2019-03-25
广东农业科学(2017年5期)2017-08-29
中国工程咨询(2015年5期)2015-02-16
