
文本挖掘在基因组注释中的应用
2017-03-21 10:50:06,
中华医学图书情报杂志 2017年3期
,
基因组注释是指利用生物信息学方法对基因组中所有基因的生物学功能进行高通量注释,包括核苷酸级别的注释、蛋白质级别的注释以及流程级别的注释[1]。目前,常规的基因组注释方法存在步骤过于繁琐、需要借助高精尖设备、人工操作存在误差、 “同源-功能相似”只是一种假说、模体本身具有的层次性以及涉及的分析工具较多无法自动化操作等问题,得到的结果存在误差[2]。随着计算机技术的发展以及关于基因研究的生物医学文献数量的不断增加,利用文本挖掘技术[3]对生物医学文献分析来实现对基因组注释成为一种新的研究趋势。
1 材料和方法
笔者利用WOS数据库中的文献作为研究的样本来源,检索策略为:TS=(gene annotation* OR genomic* annotation*) AND TS=(text mining OR literature mining),检索时间为2016年10月19日,限定时间段在2000-2016年之间,得到328篇相关文献。利用书目共现分析软件BICOMB抽取相关文献中的引文,选取出现频次在15次及以上的引文,共得到16篇高被引论文(表1)。利用BICOMB构建高被引论文——来源文献矩阵(该矩阵可反映高被引论文在来源文献中的分布情况),然后将词篇矩阵导入聚类分析软件gCluto中进行高被引论文的同被引聚类分析。

表1 328篇来源文献中的高被引论文(n=16,f>=15)
2 结果与分析
将同被引聚类分析结果用可视化图像表示,山峰图见图1,棋盘图见图2。图1中16篇高被引论文根据其在328篇来源文献中的被引情况可分成3个大类;图2中行聚类是对于高被引论文的聚类,列聚类是对于来源文献的聚类。图2中行聚类结果也表明该16篇高被引论文可分为3类,表示文本挖掘技术在基因组注释中的3个应用方向。各大类对应的高被引论文见表2。其中每个大类的内容可根据该大类中包含的高被引论文及其间的树状关系进行总结,通过对每个大类对应的列聚类中描述度较高的来源文献(即每个类的类标签文献)的阅读研究进一步把握各大类的内容。本文结合同被引论文聚类分析结果和各类中高被引论文,将文本挖掘技术在基因组注释方面的应用分为权威工具的使用、文本挖掘工具和算法的开发、文本挖掘工具的检验3类。
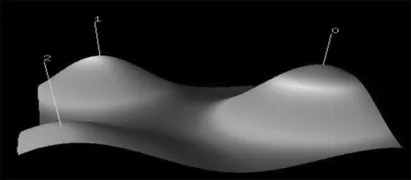
图1 高被引论文聚类分析的山峰图
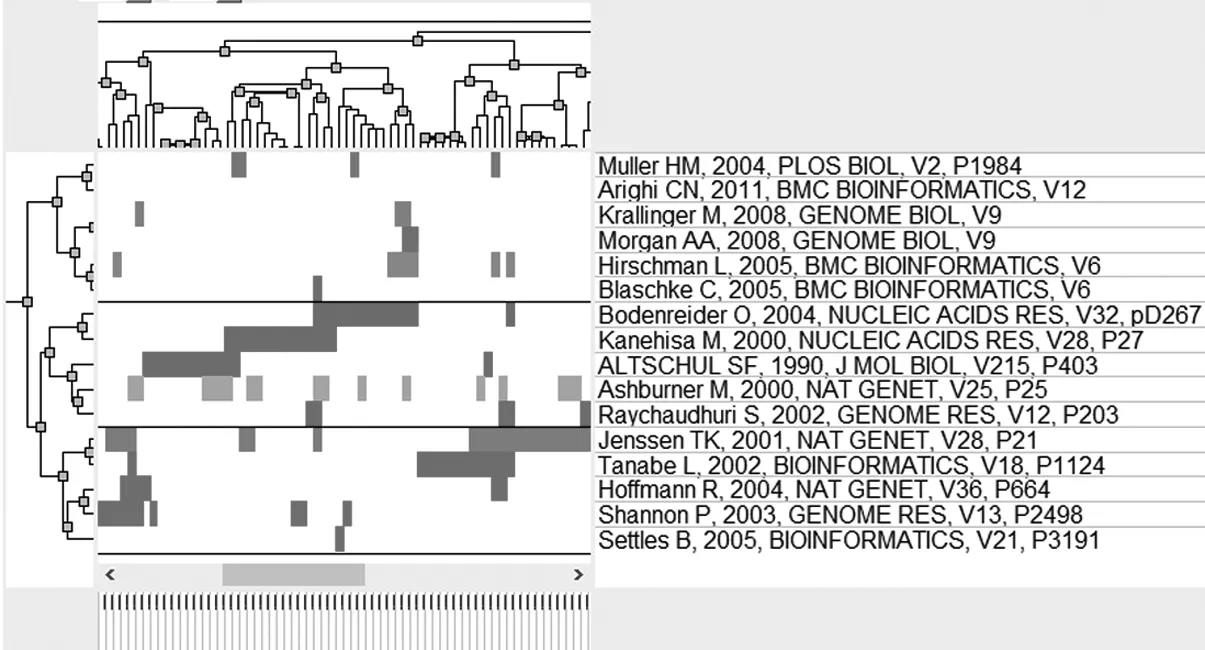
图2 高被引论文聚类分析的棋盘图

表2 3类对应的高被引论文
一是权威工具的使用。通过对Cluster 1中相关高被引文献以及类标签文献的分析,总结出在基因组注释的相关研究中,收录有基因组及基因产物相关序列、结构或功能信息的数据库和软件工具以及与基因相关的受控词汇表被广泛利用,如京都基因和基因组百科全书(Kyoto Encyclopedia of Genomes, KEGG)[4-5]、一体化医学语言系统(The Unified Medical Language System, UMLS)[6-7]、基因本体(Gene Ontology, GO)[8]、基本局域联配搜索工具(Basic Local Alignment Tool, BLAST)等。这些数据库、软件将已知的基因相关信息汇总、整理并组织起来,提供给科研人员使用和查询。Taniya T等人[9]在寻找特定复杂疾病新的候选基因的研究中利用了京都基因和基因组百科全书、基因本体以及其他一些数据库中的信息来获取与类风湿性关节炎和前列腺癌相关的已知致病基因。
然而这些数据库或软件工具中有些关于基因、蛋白质等物质的注释信息基本依赖于专家人工从文献集中获得。随着生物医学科技文献数量的增加以及用户需求的增加,这种数据收集方法缺乏灵活性,其收录信息的范围也受到限制。因此从文献中自动提取信息的计算机算法被开发出来作为人工开发数据库的补充,尤其是基因概念之间的关联研究及应用[10-11]。
二是文本挖掘工具和算法的开发。对Cluster 2中相关高被引文献进行分析,五篇高被引论文的研究方向都是对于文本挖掘工具的介绍,包括基因和蛋白质等相关实体的识别工具[12-13]、基因共现网络创建工具[14]、利用基因与蛋白作为链接点构建文献网络的信息系统[15]等等。在此基础上再对Cluster 2中的类标签文献进行分析,我们总结出在基因组注释中,相关文本挖掘工具和算法的开发与利用是文本挖掘技术在基因组注释方面的一大重要应用。
在分子生物学及相关领域,大规模高通量实验技术的发展和生物信息学工具的使用产生了大量的数据并促进了科学文献的增长,但也使得许多显性或隐性知识被掩盖在文献中难以被科研人员利用,这促进了文本挖掘工具和算法的发展与利用[16]。通过Rodriguez-Esteban R等人[17]与Krallinger M等人[18]对于生物医学领域文本挖掘技术的论述,我们可以总结出文本挖掘技术涉及到命名实体识别、关系检测、知识发现等多个阶段,在各个阶段中都有相关的文本挖掘工具或应用程序被开发出来。比如在命名实体识别阶段,有Whatizit系统(一个文本处理系统,可以识别文本中的分子生物学术语,并将其链接到公共可用的数据库中)、ABNER程序(A Biomedical Named Entity Recognizer,生物医学命名实体识别器,是一个可以识别蛋白质、DNA、RNA、细胞系和细胞类型这五种术语的开源软件工具)等工具;在关系检测阶段,有MedGene(一种全面估计和总结Medline中所有人类基因——疾病关系相对强度的文本挖掘工具)等工具,并且基因本体和蛋白质相互作用网络也能分别展示相关基因、蛋白质的亲疏远近关系;在知识发现阶段,有Arrowsmith(一个免费的、基于公共网络的两节点搜索工具,允许用户在PubMed中识别任何两组文章集之间有生物学意义的连接)等工具。
三是文本挖掘工具的检验。对Cluster 0中相关高被引文献进行分析,6篇高被引论文中有5篇文献的主要内容是对于BioCreative(Critical Assessment of Information Extraction systems in Biology,生物学中信息提取系统的严格评价)评估的描述[19-23],再结合对Cluster 0中描述度较高的类标签文献的分析,发现文本挖掘在基因组注释中的一大应用是进行文本挖掘竞赛以检验各文本挖掘工具。
在生物医学领域,已有很多关于基因、蛋白等物质的注释数据库被开发。随着生物医学领域科技文献量的增长,依靠专家人工从文献中提取有用信息策展相关数据库在时间上已经有很大的局限性,这促进了生物医学领域文本挖掘技术尤其是自然语言处理技术的发展,也使得BioCreative评估应运而生。 BioCreative评估建立于2004年,主要目的在于评估应用于生物医学领域的文本挖掘技术的最高水平。除此之外,该评估还促进了相关数据库开发者与文本挖掘研究人员之间的交流,有利于自动化的文本挖掘技术与人工策展相结合共同进行数据库的开发。 从2004年开始,BioCreative评估用来检验各文本挖掘工具的任务多围绕文献中基因、蛋白质等相关实体的提取、基因标准化、利用基因本体或蛋白质相互作用网络在全文中提取基因或蛋白质的功能注释等方面展开,在这期间还邀请文本挖掘工具最终用户参与进来,加强文本挖掘工具解决生物医学研究中实际问题的能力[18,24-26]。
3 讨论
本文通过对WOS中有关文本挖掘与基因组注释的相关文献的检索、筛选、聚类和阅读研究,发现文本挖掘技术在基因组注释方面的应用大致分为权威工具的使用、文本挖掘工具和算法的开发、文本挖掘工具的检验3方面。伴随着生物医学文献量的不断增加、高通量实验技术的不断进步以及科研人员对于信息提取工具需求的增加,相信会有越来越多的文本挖掘工具被开发出来。与此同时,随着文本挖掘工具竞赛的举办,其研发会越来越贴近科研人员的现实需要。对于依靠人工从文本集中收集有用信息的数据库等工具的研发,未来的发展趋势应该会将文本挖掘技术整合进相关开发流程,更加依赖文本挖掘技术来提取信息以充实数据库。当然,除了在基因组注释方面,文本挖掘技术在药物重定位研究、药物靶向位点研究等其他生物医学领域也会发挥越来越重要的作用。
猜你喜欢
科学与社会(2022年4期)2023-01-17 01:20:04
科学与社会(2021年4期)2022-01-19 03:29:50
今日农业(2021年11期)2021-08-13 08:53:24
小太阳画报(2020年11期)2020-12-10 06:50:08
小太阳画报(2020年10期)2020-10-30 01:57:15
图书馆建设(2018年5期)2018-07-10 09:46:44
读者(2017年18期)2017-08-29 21:22:03
照明工程学报(2016年3期)2016-06-01 12:17:54
遗传(2014年3期)2014-02-28 20:58:49
世界科学(2014年8期)2014-02-28 14:58:31
