
碱木质素基活性炭的制备与孔结构特征
2017-02-22 08:08张冠中赵师辛陈梦涵
林业机械与木工设备 2017年2期
张冠中, 赵师辛, 陈梦涵, 林 剑, 王 霁
(北京林业大学木材科学与工程北京市重点实验室,北京100083)
研究与设计
碱木质素基活性炭的制备与孔结构特征
张冠中, 赵师辛, 陈梦涵, 林 剑*, 王 霁
(北京林业大学木材科学与工程北京市重点实验室,北京100083)
为高值化利用碱木质素,本研究采用不同活化方法制备碱木质素基活性炭,通过氮气吸附-脱附和扫描电镜表征其孔隙结构和表观形貌,结果表明:①炭化处理不利于碱木质素在活化过程中孔隙的生成,未炭化处理的碱木质素基活性炭的比表面积和孔容相对较高;②氯化锌活化比水蒸气活化碱木质素可获得更高的比表面积和孔容,分别达到1 600 m2/g和1.16 cm3/g,二次活化对碱木质素基活性炭具有扩孔作用,尤其是水蒸气与氯化锌同时活化的扩孔作用更明显,使其比表面积相对减少,而孔容略微增加;③碱木质素基活性炭的孔径主要分布于超微孔0.5 ~0.6 nm、微孔0.8 ~2 nm和中孔3 ~10 nm三个区域,二次活化可使微孔减少中孔增加,并相对均匀地分布在活性炭表面。
碱木质素;活性炭;氮气吸附;孔结构
作为最重要的工业吸附剂之一,活性炭是一种具有发达孔隙结构、较高比表面积、多种表面官能团(如羧基、羰基、羟基等)、较大机械强度,以及耐酸、耐碱、耐热、失效后易再生等特性的含碳物质,被广泛应用于空气净化、废水处理、催化剂载体、电极材料等方面。目前,制备活性炭的原料主要来自于化石类资源(烟煤、褐煤、沥青等)和生物质资源(木材、果壳、竹材等)[1]。
木质素作为地球上仅次于纤维素的第二大可再生生物质资源,每年产量十分丰富,仅制浆造纸分离出来的木质素就多达5 000万t/年[2],而被有效利用的木质素却不及总分离产量的5%。这些木质素的三种单体中含有六碳环结构,其碳含量为60%~65%,是制备炭材料的理想前驱体[3]。因此,以制浆造纸分离提取的木质素为原料制备活性炭,不仅可以解决制浆造纸废液排放引起的环境污染和活性炭原料资源紧张的问题,而且还可以高值化利用木质素达到变废为宝的目的。
目前,用于制备木质素基活性炭的原料主要是磺酸盐木质素、硫酸盐木质素和酸水解木质素等[4-9],采用碱木质素制备活性炭的研究报道很少。制备木质素基活性炭主要采用的是反应温度低、比表面积和产率高的化学活化法[10],但其获得的活性炭中孔比例相对较低。本研究以碱法制浆造纸废液中分离提取的碱木质素为原料,采用不同的活化方法制备碱木质素基活性炭,通过氮气吸附测定和扫描电镜观察,研究不同活化方法对碱木质素基活性炭比表面积、孔容、孔隙分布和表面形貌等特征的影响,以高值化利用碱木质素。
1 试验材料与方法
1.1 试验材料
(1)黑液粉末:碱法制浆造纸黑液粉末购于新沂市飞皇化工有限公司,具体参数为:水分≤7.0%,pH值11~12,木质素含量约30%。
(2)盐酸,分析纯,购于北京北化精细化学品有限责任公司。
(3)氯化锌,分析纯,购于北京化工厂。
1.2 碱木质素提取
将800 g黑液粉末与4 L水混合,搅拌均匀后采用滤纸过滤,以去除黑液中的不溶物;取滤液逐次加入盐酸,直至滤液的pH值为2后采用低速离心分离机(2250 G)分离得到沉淀,再往沉淀中加入适量的水并搅拌均匀,再次离心分离,如此往复直至pH值约为5为止,将最终得到的沉淀进行冷冻干燥,即得到碱木质素粉末。
1.3 炭化处理
将碱木质素粉末置于密闭的炭化炉中,在纯度为99.9%的氮气保护下,室温保持20 min以排尽炭化炉内部空气,再以4 ℃/min的升温速率从室温加热到500 ℃并在该温度下保温1 h,经过自然冷却至室温后即可获得炭化后的碱木质素粉末。
1.4 化学活化处理
将原料与浓度为40%的氯化锌溶液按1:3的比例混合,采用磁力搅拌器搅拌2 h,在电热鼓风干燥箱中烘干后置于活化炉中,在纯度为99.9%的氮气保护下,室温保持20 min以排尽炭化炉内部空气,再以4 ℃/min的升温速率从室温加热到700 ℃并在该温度下保温1 h,经过自然冷却至室温后即可获得化学活化后的碱木质素粉末;然后使用1 mol/L的盐酸对其进行洗涤,再使用自来水将其pH值洗至中性,最后烘干得到碱木质素基活性炭。
1.5 物理活化处理
将原料置于活化炉中,在纯度为99.9%的氮气保护下,室温保持20 min以排尽炭化炉内部空气,再以4 ℃/min的升温速率从室温加热到700 ℃,当温度达到700 ℃时通入水蒸气进行活化,水蒸气流量控制在3.125 g/min,保温1 h,然后自然冷却至室温后即可获得碱木质素基活性炭。
1.6 孔隙结构检测
采用美国Quantachrome公司生产的AutosorbiQ全自动比表面积与孔径分布仪来测定活性炭的氮气吸附曲线。测定前,样品首先在300 ℃条件下脱气3 h,之后将其放在液氮环境中(77 K)进行氮气吸附。用BET(Barrett Emmett Teller)法计算样品的总比表面积[11];样品的总孔容由相对压力为0.995 时吸附的N2体积得到;用t-plot法计算其微孔比表面积和微孔孔容[12];中孔比表面积和孔容用BJH(Barrett Joyner Halenda)方法计算;用DFT(Density Functional Theory)法分析样品的全孔分布。
1.7 表面形貌观察
扫描电镜(SEM)采用日立公司生产的S-3400N型扫描电子显微镜,观察碱木质素基活性炭的表观形貌。
2 试验结果与讨论
2.1 一次活化对活性炭的影响
一次活化碱木质素制备活性炭的氮气吸附-脱附曲线如图1所示。图1中,AC-W为水蒸气活化,AC-C-W为炭化后水蒸气活化,AC-Z为氯化锌活化,AC-C-Z为炭化后氯化锌活化(下同)。
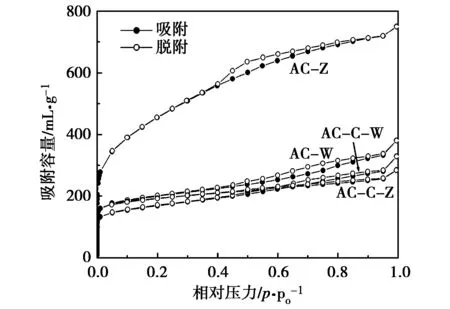
图1 一次活化制备活性炭的氮气吸附-脱附曲线
与经过炭化处理再活化的活性炭AC-C-W和AC-C-Z相比,直接活化得到的活性炭AC-W和AC-Z的氮气吸附量均有所增加,尤其是AC-Z的吸附量增加十分明显,是AC-C-Z的3倍左右,表明炭化处理会减少样品在活化过程中孔隙的生成。这可能是因为碱木质素经过炭化后,木质素分子链之间发生了聚合和交联,使结构变得更加紧密,在活化时活化剂不容易进入木质素内部,因而使木质素的活化程度降低。另外,在相对压力达到0.1以后,AC-C-W和AC-C-Z的氮气吸附量随着相对压力的增加而稍有增加,并且曲线没有出现明显的吸附滞后环,这说明样品中仍以微孔为主并存在少量的中孔[13];而AC-W和AC-Z的氮气吸附曲线均有吸附滞后环,表明存在较多的中孔,特别是AC-Z的氮气吸脱附曲线在相对压力达到0.1以后随着相对压力的增加而呈线性增大,表明AC-Z存在更多中孔。
一次活化方法制备活性炭的总比表面积和总孔容见表1。经炭化处理的AC-C-W和AC-C-Z的比表面积和孔容分别为609 m2/g与0.509 cm3/g,730 m2/g与0.440 cm3/g,均小于相应未炭化处理的活性炭;同时,氯化锌活化得到活性炭的比表面积和孔容均大于水蒸气活化得到活性炭的比表面积和孔容,特别是AC-Z的比表面积达到了1 601 m2/g,大于以往研究中其他木质素基活性炭的比表面积[14-15]。表明采用化学活化法有利于制备高比表面积和相对较小孔隙的木质素基活性炭。
表1 一次活化方法制备活性炭的总比表面积和总孔容

样品总比表面积/m2·g-1总孔容/cm3·g-1AC-W7300.590AC-C-W6090.509AC-Z16011.160AC-C-Z7300.440
2.2 二次活化对活性炭的影响
二次活化碱木质素制备活性炭的氮气吸附-脱附曲线如图2所示。图2中,AC-W+Z为水蒸气活化与氯化锌活化同时进行,AC-C-W+Z为炭化后水蒸气活化与氯化锌活化同时进行,AC-Z-W为氯化锌活化后再水蒸气活化,AC-C-Z-W为炭化后先氯化锌活化再水蒸气活化(下同)。
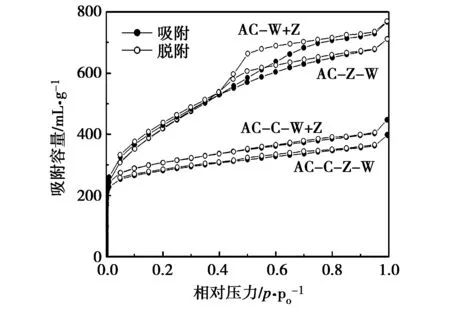
图2 二次活化制备活性炭的氮气吸附-脱附曲线
与一次活化结果类似,在二次活化中,经过炭化处理制备的活性炭AC-C-W+Z和AC-C-Z-W的氮气吸附量均小于相应的未炭化的活性炭的吸附量,再次表明炭化处理不利于制备多孔隙的碱木质素基活性炭。在相对压力达到0.1以后,AC-C-W+Z和AC-C-Z-W的氮气吸附量随着相对压力的提高稍有增加,而且曲线没有出现明显的吸附滞后环,这说明样品中仍以微孔为主并存在少量的中孔。而AC-W+Z和AC-Z-W的氮气吸脱附曲线在相对压力达到0.1以后随着相对压力的增加均呈线性增大,表明二者存在大量中孔;而且AC-W+Z的氮气吸附曲线存在的吸附滞后环明显大于AC-Z-W,表明存在更多的中孔。
二次活化方法制备活性炭的总比表面积、总孔容见表2。所有经过二次活化得到活性炭的比表面积均大于1 000 m2/g,最大值为1 513 m2/g,表明二次活化可以制备高比表面积的碱木质素基活性炭[16-19]。
表2 二次活化方法制备活性炭的总比表面积和总孔容

样品总比表面积/m2·g-1总孔容/cm3·g-1AC-W+ZAC-C-W+ZAC-Z-WAC-C-Z-W14811147151310581.1870.6931.1010.614
2.3 活性炭孔结构特征
由于在一次活化和二次活化中炭化处理均使活性炭的孔隙减少,因此选取无炭化处理的活化炭来进行其孔结构分析。不同活化方法制备的活性炭孔结构之间存在一定差异。图3所示为采用DFT方法得到的不同活化方法制备活性炭的孔径分布图,活性炭的孔径主要分布在超微孔0.5 ~0.6 nm、微孔0.8~2 nm和中孔3 ~10 nm三个区域。对于AC-W,孔径主要集中在超微孔区域,并有很少量的中孔,使其微孔比表面积较大,中孔比表面积相对较少,这也与前面其氮气吸附-脱附曲线分析一致。可能微孔分布较窄且中孔区域的孔径偏大而广,AC-W中孔孔容较大,进而引起平均孔径较大,达到3.23 nm。当采用化学活化法时,AC-Z在超微孔区域的孔容减少,而在微孔和中孔区域的孔容增加,表明化学活化法比物理活化法更容易制备具有相对较大孔隙的活性炭。不同活化方法制备活性炭微孔和中孔的比表面积、孔容以及中孔比例见表3。
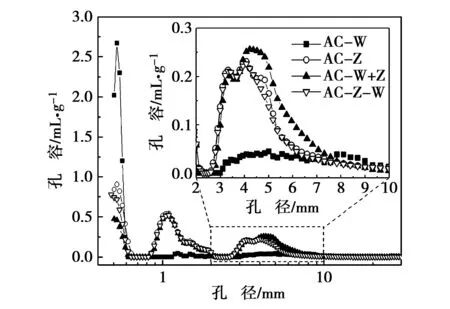
图3 不同活化方法制备活性炭的孔径分布
表3 不同活化方法制备的活性炭的微孔和中孔比表面积与孔容以及中孔比例
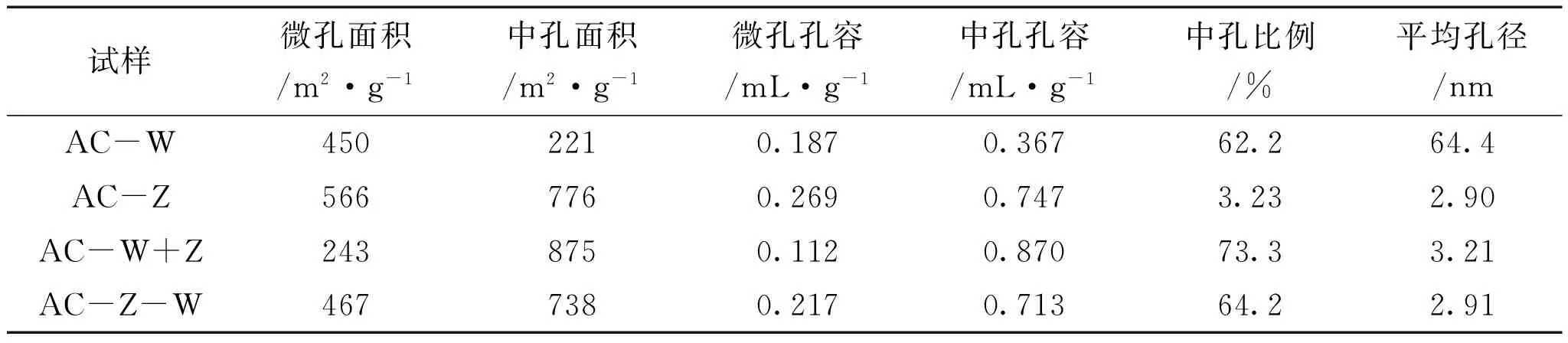
试样微孔面积/m2·g-1中孔面积/m2·g-1微孔孔容/mL·g-1中孔孔容/mL·g-1中孔比例/%平均孔径/nmAC-W4502210.1870.36762.264.4AC-Z5667760.2690.7473.232.90AC-W+Z2438750.1120.87073.33.21AC-Z-W4677380.2170.71364.22.91
采用二次活化方法时,活性炭在微孔区域的分布与一次活化时的化学活化法相近,但超微孔的孔容有所减少,与一次活化时的物理活化法相比下降明显,尤其是AC-W+Z在超微孔处的孔容最少,这可能是由于二次活化时水蒸气在氯化锌活化的基础上主要进行扩孔行为,而在微孔产生方面的活化则相对减弱,使得超微孔孔容减少,中孔孔容增加,中孔比例达到73.3%,这也是中孔区域中AC-W+Z向孔径变大方向移动的原因。另外,由于二次活化的扩孔作用,使微孔比表面积减少,中孔比表面积增加,但总比表面积仍小于一次活化的总比表面积。因此,一次活化可制备高比表面积但孔径相对较小的碱木质素基活性炭,而二次活化与之相反,即有利于制备孔径相对较大而比表面积相对较小的碱木质素基活性炭。
2.4 活性炭表观形貌
图4所示为不同活化方法条件下制备得到的碱木质素基活性炭SEM图。从图4(A)可以看到,水蒸气活化得到的活性炭的孔较少,而且主要是小孔,并多分布在活性炭表面;在图4(B)中,氯化锌活化得到的活性炭的孔较多,孔径相对较大,大小不一,并且在活性炭内部也能够观察到孔的存在,这可能是由于两种活化方法的处理方式不同造成的。水蒸气活化时,活化剂水蒸气是碱木质素被加热至高温后才开始通入,此时碱木质素在高温下部分熔化,使水蒸气较难进入碱木质素内部,而仅是在表面活化,这也是制备相应的活性炭比表面积较低的原因;而氯化锌活化时,碱木质素先与氯化锌溶液混合,由于碱木质素具有相对较松散的分子结构,使氯化锌溶液能够比较容易进入碱木质素内部,当干燥处理后,氯化锌颗粒便残留在内部,并在高温活化时能够很充分的与碱木质素接触,使得活化效果较好,比表面积较高。图4(C)和图4(D)是二次活化后碱木质素的外观形貌,与AC-W和AC-Z,AC-W+Z和AC-Z-W相比表面拥有了较多的孔,并且分布比较均匀,这主要是水蒸气和氯化锌共同活化作用的结果,表明二次活化对活性炭的表面形貌影响十分显著。

图4 不同活化方法条件下制备得到的
3 结论
(1)炭化处理不利于碱木质素在活化过程中孔隙的生成,未经炭化处理的碱木质素基活性炭的比表面积和孔容相对较高;
(2)氯化锌活化比水蒸气活化碱木质素可获得更高的比表面积和孔容,分别达到1 600 m2/g和1.16 cm3/g;二次活化对碱木质素基活性炭具有扩孔作用,尤其是水蒸气与氯化锌活化同时进行时扩孔作用更明显,使其比表面积相对减少,而孔容略有增加;
(3)碱木质素基活性炭的孔径主要分布于超微孔0.5 ~0.6 nm、微孔0.8 ~1 nm和中孔3 ~10 nm三个区域,二次活化可使微孔减少中孔增加,并相对均匀地分布在活性炭表面。
[1] 闫伟.草浆黑液木质素制备活性炭及其在油气回收中的应用[D].大连:大连理工大学,2011.
[2] Mai C,Milstein O,Huttermann A.Chemoenzymatical grafting of acrylamide onto lignin[J].Journal of Biotechnology,2000(79):173-183.[3] 唐一林,江成真,刘晓敏,等.由木质素制备颗粒活性炭的方法[P].中国专利:201310044521.2,2014.
[5] 颜涛,李云雁,宋光森,等.氯化锌法制备木质素活性炭的研究[J].安徽农业科学,2008,36(28):12094-12096.
[6] Daniel M,Vanessa T F,Vanessa F.Activated carbons from lignin:kinetic modeling of the pyrolysis of kraft lignin activated with phosphoric acid[J].Chemical Engineering Journal,2005(106):1-12.
[7] Gonzales-Serrano E,Cordero T,Rodriguez-Mirasol J,et al.Rodriguez JJ.Removal of water pollutants with activated carbons preparedfrom H3PO4 activation of lignin from kraft black liquors[J].Water Research,2004,38(13):3043-3050.
[8] Gonzalez-Serrano E,Cordero T,Rodríguez-Mirasol A J,et al.Development of porosity upon chemical activation of kraft lignin with Zncl2[J].Industrial & Engineering Chemistry Research,1997,36(11):4832-4839.
[9] Fierro V,Torne V,Montane D,et al.Study of the decomposition of kraft lignin impregnated with orthophosphoric acid[J].Thermochimica Acta,2005,433(1-2):142-148.
[10] 周建斌,张齐生,高尚愚.水解木质素制备药用活性炭的研究[J].南京林业大学学报,2013,27(5):4-6.
[11] 蒋莉,马飞,梁国斌,等.木质素活性炭的制备及工艺优化[J].新型炭材料,2011,26(5):396-400.
[12] Brunauer S,Emmett P H,Teller E.Adsorption of gases in multimolecular layers[J].Journal of the American Chemical Society,1938,60(2):309-319.
[13] Boer J H D,Lippens B C,Linsen B G,et al.The t-curve of multimolecular N2-adsorption[J].Journal of Colloid & Interface Science,1966,21(66):405-414.
[14] Lee J G,Kim J Y,Kim S H.Microtexture and electrical properties of PAN-ACF[J].Journal of Materials Science,2007,42(7):2486-2491.
[15] Hayashi J,Kazehaya A,Muroyama K,et al.Preparation of activated carbon from lignin by chemical activation[J].Carbon,2000,38:1873-1878.
[16] 田龙,马晓建.木质素基活性炭的制备和吸附特性[J].材料研究学报,2013,2(2):219-224.
[17] 魏海博.高比表面积椰壳基活性炭制备及其在超级电容器中应用研究[D].长沙:国防科学技术大学研究生院,2012.
[18] 耿新,李莉香,安百刚,等.二次活化对椰壳基活性炭电化学性能的影响[J].沈阳农业大学学报,2013,44(1):75-76.
[19] 赵赫,马尔妮,赵广杰.KOH活化木材苯酚液化物炭纤维的制备与孔结构分析[J].林业机械与木工设备,2014,42(5):22-26.
(责任编辑 张雅芳)
Preparation and Pore Features of Soda Lignin-based Activated Carbon
ZHANG Guang-zhong, ZHAO Shi-xin, CHEN Meng-han, LIN Jian*, WANG Ji
(Beijing Key Laboratory of Wood Science and Engineering,Beijing Forestry University,Beijing 100083,China)
To promote the high value-added utilization of soda lignin,soda lignin-based activated carbon(AC)is prepared with various activation methods,with pore structure and apparent morphology characterized through nitrogen adsorption-desorption and scanning electron microscopy.The results show:①carbonization is not conducive to the generation of pores of soda lignin in the activation process and the specific surface area and pore volume of the soda lignin not subjected to carbonization are relatively higher;②higher specific surface area(1600 m2/g)and pore volume(1.16 cm3/g)can be obtained through zinc chloride-based activation than vapor-based activation,and the pore enlarging role is significantly enhanced when vapor and zinc chloride are used for activation simultaneously,with the specific surface area relatively reduced,pore volume slightly increased;③the pore diameter of soda lignin-based AC is mainly distributed in three areas,i.e.0.5 nm-0.6 nm(ultro-micropores)and 0.8 nm-2 nm(micropores)as well as 3 nm-10 nm(mesopores)and secondary activation can reduce the number of micropores and increase the number of mesopore,which are relatively evenly distributed on the surface of AC.
soda lignin;activated carbon;nitrogen adsorption;porous structure
2016-10-13
北京林业大学“国家级大学生创新创业训练计划”项目(201610022057);北京市教委科学研究与研究生培养共建项目——重点学科(2015)
张冠中(1995-),男,山东济宁人,大学本科在读,主要从事木质基活性炭方面的研究。
*通讯作者:林 剑(1986-),男,福建宁德人,讲师,博士,主要从事木质基碳材料方面的研究,E-mail:linjian0702@bjfu.edu.cn。
TS612
A
2095-2953(2017)02-0035-05
猜你喜欢
能源化工(2022年3期)2023-01-15
世界有色金属(2021年6期)2021-06-14
净水技术(2020年12期)2020-02-16
无机盐工业(2019年6期)2019-12-24
中国化工贸易·中旬刊(2019年8期)2019-10-21
电子制作(2019年12期)2019-07-16
世界有色金属(2018年13期)2018-09-12
北京航空航天大学学报(2017年9期)2017-12-18
科技创新导报(2016年35期)2017-04-20
太原理工大学学报(2012年3期)2012-10-26
