
龙马溪组页岩干酪根平均分子结构模型的构建
2017-02-17 12:54:49刘向君罗丹序熊健梁利喜
化工进展 2017年2期
刘向君,罗丹序,熊健,梁利喜
(西南石油大学油气藏地质及开发工程国家重点实验室,四川 成都610500)
龙马溪组页岩干酪根平均分子结构模型的构建
刘向君,罗丹序,熊健,梁利喜
(西南石油大学油气藏地质及开发工程国家重点实验室,四川 成都610500)
以四川盆地下志留统龙马溪组下段页岩中提取的干酪根为研究对象,利用等温吸附、XRD、FTIR和13C NMR实验分析其基本的结构特性,获得了重要结构信息。研究结果表明:该地区干酪根是非晶态高聚物;通过对比干燥干酪根和平衡水干酪根的等温吸附曲线,发现平衡水干酪根的吸附能力较低,这与干酪根表面吸附的水分子有关,说明干酪根分子中含有亲水的官能团;通过FTIR对龙马溪组干酪根分析发现其含有苯环、羟基、亚甲基和甲基等官能团;13C NMR谱图表明该地干酪根由芳碳、脂碳和羰基碳构成,其中芳烃是其主要的结构成分,脂碳和羰基碳起连接芳香结构单元的作用。利用这些信息,进而建立起了龙马溪组干酪根的平均分子结构模型,最后通过对比实验结构参数和模拟模型的结构参数,可以发现两者结果接近,说明模拟的干酪根分子模型是可以用于下一步干酪根吸附性能的分子动力学模拟。
龙马溪组;页岩;干酪根;平均分子结构
随着环境污染严重和能源技术开发的发展,人们对非常规能源的开发越来越关注,特别是非常规页岩气。根据美国能源信息署2015年最近更新的世界页岩资源评估报告指出全球页岩气的技术可采资源总量为214.55×1012m3,其中中国的技术可采资源储量为31.6×1012m3[1],说明我国的页岩气资源丰富。因此,页岩如果能成功地进行商业化开发开采可以极大地缓解我国的能源压力,改变能源结构。
页岩气的赋存状态有游离态、吸附态和溶解态[2]。CURTIS[3]指出美国页岩气藏中20%~85%的是吸附气,说明吸附气在页岩气储量中占有重要地位。根据目前研究可知,影响页岩的吸附性能的因素有很多,主要有有机质、矿物组成、温度、压力和含水量等。CHALMERS等[4]和闫建萍等[5]研究都表明页岩中的有机质(TOC含量、干酪根类型和有机质成熟度)能影响页岩吸附甲烷的能力。薛海涛等[6]发现干酪根对甲烷的吸附量远大于泥岩和灰岩。薛华庆等[7]也提出页岩中比表面积和吸附能力的主要贡献者是干酪根。GUO等[8]指出页岩中提取的干酪根的吸附量最大,页岩的吸附量次之,页岩中提取干酪根后剩余的矿物吸附量最小。2012年,ZHANG等[9]发现干酪根类型对页岩吸附气量的影响顺序为:typeⅠ>typeⅡ>typeⅢ。上述研究都说明页岩中有机质对甲烷的吸附性能有重要影响。
目前研究干酪根对甲烷吸附的影响的方法主要有物理实验法和数值模拟法。物理实验方法主要是等温吸附实验,而数值模拟方法是近几年才发展起来的分子动力学方法。物理实验往往无法模拟地层中高温高压下干酪根吸附甲烷的状态,也无法得出干酪根吸附的微观机理;分子动力学法可以解决物理实验的这些缺陷。而利用分子动力学方法研究干酪根对甲烷吸附性能的时候,往往采用的是干酪根的近似模型,进而使用相关巨正则蒙特卡罗方法进行模拟研究。例如,欧志鹏[10]采用的是石墨烯片层结构来研究甲烷在有机层状空隙中的赋存与扩散;MOSHER等[11]也是采用理想的石墨模拟单元进行煤和页岩系统中微孔和中孔的甲烷吸附分子模拟;2014年,FIROUZI等[12]在石墨结构的基础上,利用孔径分布和孔隙度数据来获得了更逼真的煤和页岩中有机质的模拟模型;同年,FIROUZI研究团队[13]又利用该模型来研究了氮气的滑脱效应,从而更加准确地预测了页岩中气体的运移状态;石墨结构也被WANG等[14]用来模拟研究页岩有机质孔隙和狭缝中液态链烃的赋存状态;刘冰等[15]采用5层石墨狭缝结构来研究甲烷气体在页岩纳米微孔中的吸附现象;LIU等[16]同样也利用石墨烯结构探索了CO2在页岩气和煤中的吸附情况。这种近似结构不能有效代表干酪根的分子结构模型,利用干酪根这种近似结构研究其吸附性能是不准确的,对干酪根吸附规律的认识也会有一定偏差。
干酪根是指不溶于非氧化型酸、碱和非极性有机溶剂的分散有机质,它是页岩中有机质的主体,因此研究干酪根的分子结构对评价页岩气的吸附性能有重要影响。随着研究的深入,越来越多的学者意识到如果想要更加清楚地认识到干酪根吸附气体的情况,必须建立其近似结构。虽然干酪根是一种混合物,没有一种固定的化学结构,但是可以建立干酪根的“平均分子结构”,利用其“平均分子结构”进行分子模拟的效果也一定优于之前的近似结构。“平均分子结构”是指将结构中的芳簇数进行平均后的原子集合体,不能把它当作全体中任何一个具体分子的化学结构或者分子的基本单元,而是将其视为数学模型的一种,并借此反映混合物分子群体的典型化学与物理性质[17]。这种方式已经在煤、油页岩化学结构的研究中广泛使用。例如茹鑫[18]借助其建立了桦甸地区生油母质的分子模型;郑仲[19]构建了神东煤镜质组的大分子模型;王三跃[20]也在其论文中分析出了霍林河和义马褐煤的大分子结构;CANTOR[21]和QI等[22]分别分析了6种煤衍生液的平均分子结构和吸附氧过程中沥青质的平均分子结构参数变化;石油沥青质的热重力分析也是利用了其平均化学结构的核磁共振谱图来进行[23]。由此可见,使用“平均分子结构”来表征干酪根混合物的结构既是可行也是合理的。本文将利用XRD、FIRT、13C-NMR等先进技术分析并表征出该地区干酪根的“平均分子结构”模型,为下一步研究该地区干酪根的吸附特性作准备。
1 实验样品和方法
1.1 实验样品
本文中所用的页岩取自某页岩气井井下样品。同时参照《沉积岩中干酪根分离方法(GB/T 19144—2010)》制取实验所需的A、B两组干酪根。根据德国莱卡公司制造的DM4500P+QDI308型镜质组反射率测定仪的数据,可知该地区干酪根的镜质体反射率(Ro)约2.8%,属于过成熟阶段,与王哲和窦菲菲等[24-25]的研究一致。甘辉[26]研究发现该地区龙马溪组干酪根主要是Ⅰ型和Ⅱ型干酪根。通过显微组分测定实验发现该地区干酪根颜色为黑色,不透明,边缘呈棉花状,大部分为腐泥组,含量达86.3%;另外,还含有少量板状分布的、不透明的沥青质体,整体上属于Ⅱ型干酪根。有机碳实验测试结果表明该地区页岩样品的有机碳含量为3.65%,而经过处理酸处理后的得到的干酪根样品的有机碳含量为78.5%,有机硫含量为4.2%,说明干酪根平均分子模型中含有硫元素和碳元素,其中碳元素含量较高。
等温吸附实验中干酪根的平衡水处理的方法为:干酪根粉末样品被放置于装有饱和KCl溶液的30℃恒温箱内,该溶液可以使样品的相对湿度保持在96%~97%,48h后样品即被全部湿润,间隔一定时间称重一次,直到样品重量恒定为止,则达到平衡湿度。
红外实验中的样品处理方法为将2mg干酪根浓缩物和200mg溴化钾研细混合,再取60mg混合物在油压机上压成透明薄片,并在60℃通氮气条件下干燥6h。
1.2 实验仪器
(1)X射线衍射分析(XRD) 实验所用的衍射仪是由荷兰Panalytical分析仪器公司生产研发的X Pert PRO MPD,测试2θ测量范围为0~167°;步长为0.0001°。
(2)等温吸附实验 采用德国RUBOTHERM公司ISOSORP-HP型等温测量吸附仪进行,此仪器为磁悬浮天平重量法高温高压等温吸附仪,其测量的原理为重量法吸附测量法,采用恒温温度浴控温。该仪器的最高吸附实验压力和最高吸附实验温度分别为70MPa和150℃。本论文中的干酪根和平衡水干酪根的等温吸附实验是在60℃和20MPa的最高实验压力下进行的。
(3)红外光谱分析仪(FTIR) 该项实验是在由北京瑞利分析仪器有限公司制造的WQF520型傅里叶红外光谱仪上进行的,主要采用的是压片法,该仪器波数范围在7000~400cm–1,波数精度在±0.5cm–1,最优的分辨率为0.5cm–1,信噪比为10000∶1。
(4)固体核磁分析(13C NMR) 该项测试是在山西煤炭化学研究所中的Bruker AVANCEⅢTM-600MHz超导核磁共振波谱仪上完成的,采用4mm MAS探针ZrO2转子,转速为9kHz,循环延迟时间3s,碳氢交叉极化接触时间4ms。
2 实验结果与讨论
2.1 XRD结果分析
图1是干酪根样品B的XRD谱图曲线。由图1可知,干酪根不具有三维有序的晶体结构,而是非晶态高聚物,因此,从衍射图上不能获得晶体结构那样准确的结构信息。但是在2θ=26°处有一个很明显的弥散衍射峰包,称为d002峰,主要代表芳碳结构,该处信号峰较强,说明该地干酪根中芳族碳的含量较多;同时从该谱图中可以看出,在原样品中还残存着黄铁矿(Py),这是由于从页岩中提取的干酪根不可能完全去除黄铁矿。
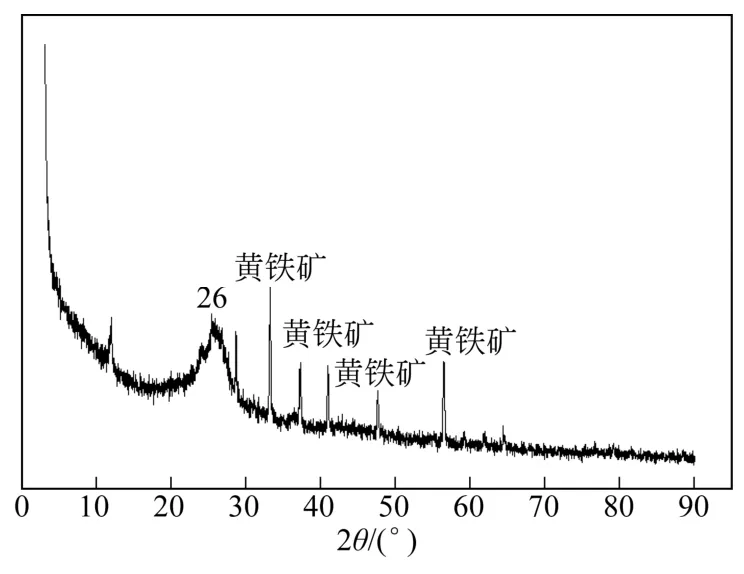
图1 龙马溪组干酪根XRD曲线
2.2 等温吸附实验结果
本实验以样品B为研究对象,干燥的干酪根和经过平衡水处理后的干酪根样品的吸附等温线见图2。从图2中可以看出,干燥干酪根的甲烷过剩吸附量低压下增加较快,而高压下降低较慢;平衡水干酪根的过剩吸附量同样也随着压力的增加先快速上升后再缓慢下降,升高和下降的速率都小于干燥的干酪根样品。但是整体上来看,平衡水干酪根的甲烷过剩吸附量要小于干燥干酪根的过剩吸附量,这说明干燥的干酪根表面可吸附一定数量的水分子,而吸附的水分子占据了一部分甲烷的吸附空间,从而导致干酪根对甲烷吸附能力的下降。这种现象可能是由于干酪根分子结构中含有某些含氧官能团,其吸附水分子而产生的,也就意味这干酪根的分子结构中存在亲水的含氧官能团。
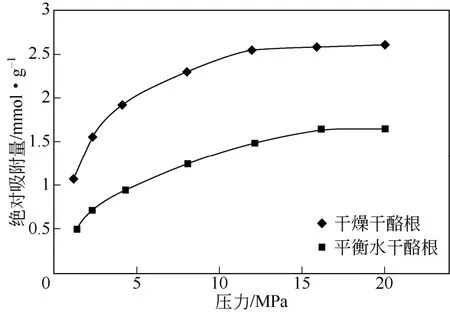
图2 干燥干酪根和平衡水干酪根的吸附等温线
2.3 红外光谱图解析
研究FTIR谱图中红外吸收峰的位置和相对强度是确定干酪根中化学基团的组成、键合性质和相对丰度的有力手段。不同基团的不同振动方式会产生不同的吸收峰,而且总是出现在一定的波段范围内;而不同峰的位置、强度和形状指示着不同的分子结构。因此,可以根据这些特征,判定出该种化合物可能含有的基团。红外光谱图上1333~4000cm–1基团特性明显,易于辨认,因此将其称为特征谱带区,这些特征峰的形状、强度和位置受其他结构的影响较小,数目也不多。另外,犹如人的指纹,特性较差,不易于辨认的1333~650cm–1则是指纹区,该区特征峰的形状复杂。
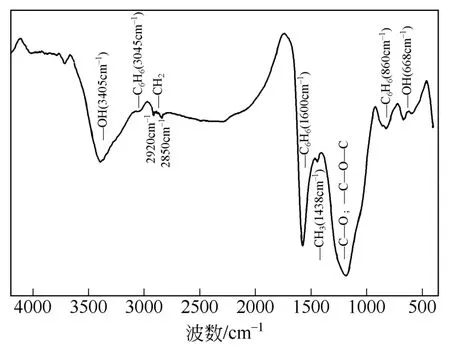
图3 龙马溪组干酪根红外光谱图
图3是样品B的红外光谱图,其波数范围主要在4500~250cm–1。在制备干酪根的过程中,往往很难完全除去其他矿物,如石英、伊利石、高岭石等硅酸盐类矿物,因此这些黏土矿物的存在会对干酪根的红外光谱图产生一定的干扰,这是无法避免的。根据光谱图,可以看出其中主要的谱带有:3404cm–1,668cm–1,1720cm–1,1180cm–1(氧原子结构);2920cm–1,2850cm–1,1460cm–1(饱和烃结构);3045cm–1,1600cm–1,860cm–1(芳香烃结构);说明干酪根的微观结构主要有芳香烃结构、饱和烃结构和含氧官能团这三部分组成。
2.3.1 烷烃类分析
2920cm–1和2850cm–1分别是脂肪族亚甲基(CH2)的反对称和对称伸缩频带,而1438cm–1是脂肪族甲基(CH3)的不对称弯曲振动和亚甲基(CH2)的剪式振动频带的综合作用,这说明该地区干酪根的分子中含有—CH2和—CH3的官能团。
2.3.2 芳香烃类分析
1600cm–1是芳环的特征吸收带,当分子的对称性增强时,该吸收带会减弱,而该吸收峰很强,说明该分子的对称性很弱,同时该谱图的强峰出现在1600cm–1处,说明该地区干酪根主要以芳烃碳为主。3045cm–1是芳核上(C—H)的伸缩振动吸收峰;860cm–1是芳氢的面外弯曲振动频带;因此,地区干酪根的分子中主要官能团就是芳香结构,这与XRD谱图的分析结果一致。
2.3.3 氧原子类分析
3404cm–1附近很宽的吸收峰是羟基(O—H)的伸缩振动频带;由前文可知干酪根可以吸水,而3404cm–1的吸收峰表示其含有亲水的羟基,因此羟基是结晶水和吸附水的表现,这和等温吸附实验结果一致;同时,在1720cm–1附近是酯、酮、酸、醛中伸缩振动吸收频带;668cm–1是羟基的面外弯曲振动频带;1180cm–1特征峰证明结构中有醚基(C—O或C—O—C)或杂原子基团(C—N、等)的存在。所以,干酪根分子中含有一些含氧官能团。
2.413C NMR谱图解析
图4是龙马溪组不同取样点A、B的两组干酪根的13C NMR谱图。从中不难发现该谱图都是由δ=0~90的脂碳区、δ=90~165的芳碳区和δ=165~220的羰基碳区三部分组成;芳碳区峰面积最大,脂碳区和羰基碳区面积较小;由此说明,芳碳是干酪根结构的主要组成部分,脂肪碳和羰基碳含量相对少些,这其中脂肪碳的作用是联接芳香结构单元;这些结论与之前红外光谱的实验结果相一致。由于该A、B两个样品的固体13C NMR谱图十分接近,因此本文作者课题组以样品B为研究对象对分子结构信息进行分析。根据干酪根的13C NMR相关文献[27],在表1中列出了干酪根中13C NMR的碳化学位移归属。

图4 A、B取样点干酪根13C NMR谱图
由表1可以得知脂肪族碳主要来源于芳甲基、脂甲基、亚甲基、次甲基、季碳、次甲基碳等官能团,而芳香族碳主要来源于质子化芳香碳、桥头芳香碳、侧支芳香碳、氧取代芳香碳等官能团。由于页岩中干酪根化学结构的复杂性和固体核磁的技术缺陷,为了获得很多干酪根化学结构的信息,对其13C NMR相关谱图进行合理的分峰拟合。根据表1中各种碳结构的化学位移归属,使用OriginPro 8.5对龙马溪组页岩中干酪根的13C NMR谱图进行分峰拟合,拟合结果见图5。由于在实验过程中产生了主峰(124峰)的魔角一级、二级旋转边带,它们是对称的出现在主峰的两侧,与主峰的距离是9000/151=59.6,为了省去边带效应的影响,所以在拟合过程中在各种特征峰的基础上,多加入了对称的两组代表旋转边带的特征吸收峰,位置分别在64和183。从图5中,还可以发现两个次高峰当中一定还含有一些碳结构,因为如果光是边带效应的话,两个次高峰的形状大小应该完全相同,从图上可以发现两者的高度和宽度都是不一致,所以两个次高峰是边带效应和若干反映一些碳结构的峰的叠加峰。此外在拟合过程中主要采用的是高斯峰形进行拟合。拟合后将各个分峰的面积和相对面积列在表1中。

图5 龙马溪组页岩中干酪根的13C NMR谱图分峰拟合结果图

表1 干酪根13C NMR分峰拟合各吸收峰的参数
从表1还可以看出64和183处的边带的面积分别为168.4和174.5,两者的差距较小,可以基本认为是位置和形状都是对称的,而在计算相对面积的时候,省去了边带效应的两个对称峰。分析表1可以得出以下结论:在该干酪根结构中,每100个碳原子中,就约有10个终端甲基碳,3个与终端甲基相连的亚甲基碳,也就是说,10个终端甲基碳中有7个非链脂甲基。脂族叔碳和季碳没有;脂族与氧相连的脂碳约有13个;5个芳环上的甲基碳,即4个与苯环相连的α与β位碳原子归属于脂链中的刚性相;11个氧接芳碳相邻的芳碳;24个质子芳碳;6个桥接芳碳;5个烷基取代芳碳;1个氧接芳碳;12个羧基碳和9个羰基碳。
计算龙马溪组页岩干酪根结构参数步骤如下。
(1)烷基碳率 烷基碳率定义为脂族碳在总碳量中的百分比。


(2)芳碳率 芳碳率定义为芳构碳在总碳量中的百分比。由于芳香族化合物的化学位移在固体13C NMR谱图上是90~165,所以常常根据谱图中90~165的特征峰的面积与0~165的特征峰的面积的比值来代表芳碳率,见式(2)。从芳碳中的碳数量几乎占到了全部碳数的一半,也就是说每100个碳原子中有8个苯环或者4个双芳环型,以此类推其他相似结构。在杨万里等[28]研究的基础上,李振广等[29]研究发现fa和H/C间有较好的相关性,当在fa<0.50时,说明该干酪根是Ⅱ。根据上面计算的芳碳率,可以确定该地区的干酪根是Ⅱ。同时,还可以发现该地区龙马溪组干酪根的分子结构的主体是芳香结构。
(3)亚甲基链的碳数 亚甲基链的碳数定义为亚甲基碳和终端芳核数的比。

由此可以推算出该地区的干酪根结构中没有亚甲基链。
(4)芳香桥碳和周碳之比XBP芳香桥碳和周碳之比XBP是重要的参数,可以用它来计算芳香簇尺度[30]。

(5)烷链支化度BI烷链支化度BI是脂族叔碳和季碳占脂碳的百分率。

说明该干酪根分子中连接芳香结构的应为长直链,而不是脂环或者支链结构。
2.5 构建干酪根平均分子结构
利用Chemdraw软件,根据之前实验信息和13C NMR谱图的解析模拟的信息,建立起该地区干酪根的平均分子结构的初始模型,然后在导入gNMR软件预测其核磁谱图,并与实验的13C NMR谱图进行对比,当gNMR软件模拟的干酪根平均分子结构与实验的谱图相吻合时,此时的干酪根的平均分子结构就是该地区的干酪根结构模型;否则就继续调整干酪根结构模型,直到模拟13C NMR谱图与实验13C NMR谱图近似为止。根据这个原理,可以确定出干酪根的的基本结构,不同的碳结构和含氧、氮或硫的结构在模拟的核磁谱图上显示的值不同;而且其含量不同,模拟谱图对应的峰的峰值会变化,含量多峰值就高,含量少峰值就低。当构建合适的结构后,gNMR软件得出的模拟谱图和实验谱图大体一致时,就可以得出合适的干酪根分子结构。根据该步骤,经过修正后的龙马溪组页岩干酪根分子模型13C NMR谱图与实验13C NMR谱图对比结果见图6。

图6 实验13C NMR谱图和模拟13C NMR的对比结果
从图6可以看出,模拟谱图和实验谱图在0~50ppm和80~170ppm有较好的拟合程度;但在50~75ppm和170~200ppm这两个区域实验谱图和模拟谱图存在一定的偏差,其原因是魔角一级、二级旋转边带的存在。实验中由于仪器的原因在主峰两侧产生面积相同的旋转边带,但是在模拟过程中谱图中没有旋转边带存在,所以两个区域的实验谱图和模拟谱图存在一定偏差。但是根据图上这两个区域的积分面积可以看出在170~200ppm中实验谱图的积分面积为0.57,而模拟谱图积分面积为0.22,两者相差0.35;而50~75ppm的区域内实验谱图的积分面积为0.37,而模拟谱图积分面积为0.02,两者相差同样也是0.35,所以50~75ppm和170~200ppm这两个区域螺旋边带的积分面积是一样的,这与实际情况中螺旋边带的性质一致,所以这两个区域的模拟谱图是合理的。
最终确定龙马溪组干酪根的分子结构如图7。从中可以得到这个分子模型主要以芳香烃为主,烯烃和羰基则是链接各个芳香烃结构单元,且主要以长直链为主。含有苯环、羟基、亚甲基、甲基、亲水的羟基等官能团,并确定其平均分子结构的化学式为C206H158O19N4S4。

图7 干酪根平均分子结构示意图
2.6 模拟结果验证
模拟的干酪根的平均分子结构的化学式为C206H158O19N4S4。根据显微组分测试可知,该地区干酪根是Ⅱ型干酪根;根据van Krevelen图版,干酪根可以分为Ⅰ型、Ⅱ型、Ⅲ型和Ⅳ型,计算的目标点到那条曲线的距离最小,该目标点就是几型干酪根[31]。而模拟结果计算出O/C为0.092,H/C为0.77,结合van Krevelen图版(图8),从图8可以发现,该O/C和H/C的值是落在Ⅱ型干酪根的区域内的红线交接处,该点离Ⅱ型干酪根的分界线比Ⅲ型干酪根的分界线更近,这说明该地区提取的干酪根是Ⅱ型干酪根,这和实验结果是一致的。再者,根据先前等温吸附实验结果,可以看出该地区的干酪根分子中是存在亲水的含氧官能团,而模拟的分子模型中—OH结构为亲水基团,与实验结果相符。另外,先前的有机碳实验表明该地区碳的含量为78.5%,有机硫含量为4.3%,而模拟结果计算出碳的含量为79.28%,硫的含量为4.1%,与实验结果较为一致。另外,实验核磁谱图中的一些结构参数和模拟结构的结构参数的对比结果见表2。
从表2中,可以看出该模拟结构的结构参数和实验的结构参数有较高的吻合度,两者的所有结构参数差别很小,说明该模拟结构是和实验相符的,该模拟结构可以为下一步模拟研究干酪根的吸附性能作铺垫。

图8 干酪根类型四分法
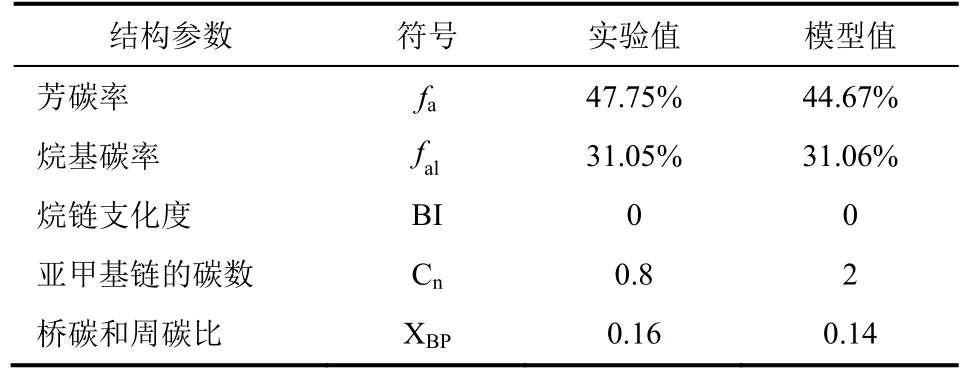
表2 实验结构参数与模拟结构参数的对比表
3 结论
(1)通过等温吸附实验结果,可知长宁地区龙马溪组页岩提取的干酪根结构中存在着亲水含氧官能团,吸附水分子后,水分子会占据一部分甲烷分子的吸附空间,从而导致其吸附甲烷的能力降低。
(2)XRD、FTIR、13C NMR三个实验所得结果一致。该地区干酪根是非晶态高聚物,含有苯环、羟基、亚甲基和甲基等官能团,其结构主要有芳香碳、脂肪碳和羰基碳构成,芳香结构是其主要成分,脂肪碳和羰基碳含量相对少些,两者的主要作用是联接芳香结构单元。
(3)根据其基本结构特性,表征出四川盆地龙马溪组干酪根的平均分子结构的化学式为C206H158O19N4S4,并建立了平均分子结构模型。比较实验结构参数和模拟的分子模型的结构参数,可以发现两者较接近,所建立的分子模型也是可行的,可以用于下一步干酪根吸附性能的分子动力学模拟。
致谢感谢中国科学院山西煤炭化学研究所为本文作者课题组提供固体13C核磁测试实验。
[1] U.S. Energy Information Administration(EIA). Technically recoverable shale oil and shale gas resources: an assessment of 137 shale formations in 41 countries outside the united states [EB/OL]. [2015-09-24]. http://www.eia.gov/analysis/studies/worldshalegas/.
[2] 张金川,金之钧,袁明生. 页岩气成藏机理和分布[J]. 天然气工业,2004,24(7):15-18. ZHANG J C,JIN Z J,YUAN M S. Reservoiring mechanism of shale gas and its distribution [J]. Natural Gas Industry,2004,24(7):15-18.
[3] CURTIS J B. Fractured shale-gas systems[J]. AAPG Bulletin,2002,86(11):1921-1938.
[4] CHALMERS G R,BUSTIN R M. Lower Cretaceous gas shales in northeastern British Columbia,Part I: geological controls on methane sorption capacity[J]. Bulletin of Canadian Petroleum Geology,2008,56(1):1-21.
[5] 闫建萍,张同伟,李艳芳,等. 页岩有机质特征对甲烷吸附的影响[J]. 煤炭学报,2013,38(5):805-811. YANG J P,ZHANG T W,LI Y F,et al. Effect of the organic matter characteristics on methane adsorption in shale [J]. Journal of China Coal Society,2013,38(5):805-811.
[6] 薛海涛,卢双舫,付晓泰,等. 烃源岩吸附甲烷实验研究[J]. 石油学报,2003,24(6):45-50. XUE H T,LU S F,FU X T,et al. Experimental study on adsorbability of methane in source rock[J]. Acta Petrolei Sinica,2003,24(6):45-50.
[7] 薛华庆,王红岩,刘洪林,等. 页岩吸附性能及孔隙结构特征——以四川盆地龙马溪组页岩为例[J]. 石油学报,2013,34(5):826-832. XUE H Q,WANG H Y,LIU H L,et al. Adsorption capability and aperture distribution characteristics of shales: taking the Longmaxi formation shale of Sichan Basin as an example [J]. Acta Petrolei Sinica,2013,34(5):826-832.
[8] GUO H,JIA W,PENG P,et al. The composition and its impact on the methane sorption of lacustrine shales from the Upper Triassic Yanchang Formation,Ordos Basin,China[J]. Marine and Petroleum Geology,2014,57:509-520.
[9] ZHANG T,ELLIS G S,RUPPEL S C,et al. Effect of organic-matter type and thermal maturity on methane adsorption in shale-gas systems[J]. Organic Geochemistry,2012,47:120-131.
[10] 欧志鹏. 纳米孔隙中甲烷扩散的分子动力学研究[D]. 成都:西南石油大学,2014. OU Z P. Molecular kinetics of methane in the nanometer pores [D]. Chengdu:Southwest Petroleum University,2014.
[11] MOSHER K,HE J,LIU Y,et al. Molecular simulation of methane adsorption in micro-and mesoporous carbons with applications to coal and gas shale systems [J]. International Journal of Coal Geology,2013,109:36-44.
[12] FIROUZI M,RUPP E C,LIU C W,et al. Molecular simulation and experimental characterization of the nanoporous structures of coal and gas shale[J]. International Journal of Coal Geology,2014,121:123-128.
[13] FIROUZI M,ALNOAIMI K,KOVSCEK A,et al. Klinkenberg effect on predicting and measuring helium permeability in gas shales[J]. International Journal of Coal Geology,2014,123:62-68.
[14] SEN W,QI H F,MING Z H A,et al. Molecular dynamics simulation of liquid alkane occurrence state in pores and slits of shale organic matter[J]. Petroleum Exploration and Development,2015,42(6):844-851.
[15] 刘冰,史俊勤,沈跃,等. 石墨狭缝中甲烷吸附的分子动力学模拟[J]. 计算物理,2013, 30(5):692-699. LIU B,SHI J Q,SHEN Y,et al. Molecular kinetics of methane in the graphite slits [J]. Chinese Journal of Computation Physics,2013,30(5):692-699.
[16] LIU Y,WILCOX J. CO2adsorption on carbon models of organic constituents of gas shale and coal [J]. Environmental Science & Technology,2010,45(2):809-814.
[17] BERKOWITZ N. The chemistry of coal [M]. Elsevier,1985.
[18] 茹鑫. 油页岩热解过程分子模拟及实验研究[D]. 长春:吉林大学,2013. XIN R. Study on the experiment and molecular simulation of oil shale pyrolysis [D]. Changchun:Jilin University,2013.
[19] 郑仲. 神东煤镜质组结构特征及其对CH4,CO2和 H2O吸附的分子模拟[D]. 太原:太原理工大学,2009. ZHEN Z. Molecular simulation study of the structure of Shendong vitrinite and the adsorption of CH4,CO2and H2O [D]. Taiyuan:Taiyuan University of Technology,2009.
[20] 王三跃. 褐煤结构的分子动力学模拟及量子化学研究[D]. 太原:太原理工大学,2004. WANG S Y. Study of lignite structure by molecular dynamics simulation and quantum chemistry [D]. Taiyuan:Taiyuan University of Technology,2004.
[21] CANTOR D M. Nuclear magnetic resonance spectrometric determination of average molecular structure parameters for coal-derived liquids[J]. Analytical Chemistry,1978,50(8):1185-1187.
[22] QI Y,WANG F. Study and evaluation of aging performance of petroleum asphalts and their constituents during oxygen absorption. III. Average molecular structure parameter changes[J]. Petroleum Science and Technology,2004,22(3/4):275-286.
[23] DONG X G,LEI Q F,FANG W J,et al. Thermogravimetric analysis of petroleum asphaltenes along with estimation of average chemical structure by nuclear magnetic resonance spectroscopy[J]. Thermochimica Acta,2005,427(1):149-153.
[24] 窦菲菲. 川东龙马溪组下部页岩储层特征研究[D]. 北京:中国矿业大学,2014. DOU F F. Reservoir characteristics research of the lower Longmaxi formation shale in East of Sichuan[D]. Beijing:China University of Mining,2014.
[25] 王哲,李贤庆,张吉振,等. 四川盆地不同区块龙马溪组页岩气地球化学特征对比[J]. 中国煤炭地质,2016,28(2):22-27. WANG Z,LI X Q,ZHANG J Z,et al. Longmaxi formation shale gas geochemical features comparison between different blocks in Sichuan Basin[J]. Coal Geology of China,2016,28(2):22-27.
[26] 甘辉. 长宁地区龙马溪组页岩气资源潜力分析[D]. 成都:西南石油大学,2015. GAN H. Shale gas resource potential analysis in Longmaxi formation of Changning district [D]. Chengdu:Southwest Petroleum University,2015.
[27] TREWHELLA M J,POPLETT I J F,GRINT A. Structure of green river oil shale kerogen: determination using solid state13C NMR spectroscopy [J]. Fuel,1986,65(4):541-546.
[28] 杨万里,李永康,高瑞祺,等. 松辽盆地陆相生油母质的类型与演化模式[J]. 中国科学,1981(8):1000-1008. YANG W L,LI Y K,GAO R Q,et al. The type and evolution mode of Terrestrial kerogen in Songliao basin[J]. Science China,1981(8):1000-1008.
[29] 李振广,秦匡宗. 用13CNMR CP/MAS波谱表征干酪根的性质[J].石油学报,1990,11(4):25-32. LI Z G,QIN K Z. Nature of kerogen characterized by solid state13C NMR spectroscopy[J]. Acta Petrolei Sinica,1990,11(4):25-32.
[30] SOLUM M S,PUGMIRE R J,GRANT D M. Carbon-13 solid-state NMR of Argonne-premium coals [J]. Energy & Fuels,1989,3(2):187-193.
[31] 熊德明,马万云,张明峰,等. 干酪根类型及生烃潜力确定新方法[J]. 天然气地球科学,2014,25(6):898-905. XIONG D M,MA W Y,ZHANG M F,et al. New method for the determination of kerogen type and the hydrocarbon potential [J]. Natural Gas Geoscience,2014,25(6):898-905.
Construction of the average molecular modeling of the kerogen from the Longmaxi formation
LIU Xiangjun,LUO Danxu,XIONG Jian,LIANG Lixi
(State Key Laboratory of Oil and Gas Reservoir Geology and Exploitation,Southwest Petroleum University,Chengdu 610500,Sichuan,China)
Taking the kerogen from the lower segment of the Longmaxi formation in Sichuan Basin as the study objects,some essential structural characteristics are obtained based on analysis of isothermal adsorption,XRD,FTIR and13C NMR. XRD analysis confirms that the kerogen exists with the long-range disordered state of aggregation. Through the adsorption isothermal experiments of the dry kerogen and equilibrium water kerogen,it is easily found that the kerogen can adsorb the water molecules and the chemical structure of the kerogen must contain hydrophilic oxygen-containing functional groups because the methane adsorption capacity of kerogen decreases after the dry kerogen adsorbed the water molecules. Adopted the FTIR to qualitatively analyze the functional groups in kerogen from Longmaxi formation,it is noted that this type of the kerogen contains the benzene ring,the hydroxyl,the methylene,the methyl and so on. Taken advantage of13C NMR to express the chemical structure of carbon atoms in kerogen,the aromatic carbon,the aliphatic carbon and the carbonyl carbon can be found there. The main structure component is the aromatic carbon,while the aliphatic carbon and the carbonyl carbon just serve as the connections among the aromatic constitutional units. In addition,the initial molecular model of kerogen isachieved based on these information. And then,the gNMR software is helpful to predict the NMR spectrum of the initial molecular model. The predicted NMR spectrum is in contrast with the experimental one. Thus,the molecular model of kerogen structure is amended until both of them are very close to gain the correct molecular model of kerogen. Meanwhile,some experimental structure parameters are acquired by the previous experiments which can compare with the structure parameters from the simulative molecular model. The contrast analysis results show that the simulative structure parameters conform with the experiment ones,indicating that the simulative molecular model can serve as the model simulated adsorption performance of kerogen from the Longmaxi formation in Sichuan Basin .
Longmaxi formation;shale;kerogen;average molecular structure
P618.13
:A
:1000–6613(2017)02–0530–08
10.16085/j.issn.1000-6613.2017.02.019
2016-07-28;修改稿日期:2016-10-25。
国家自然科学基金联合基金重点项目(U1262209)及国家自然科学基金青年科学基金项目(41602155)。
刘向君(1969—),女,教授,博士生导师,主要从事岩石力学及岩石物理、页岩气开发等方面的研究与管理工作。联系人:熊健,讲师,主要从事非常规页岩气吸附性能、岩石物理等方面的研究。E-mail:361184163@qq.com。
猜你喜欢
中学化学(2024年5期)2024-07-08 09:24:57
地球化学(2022年6期)2023-01-03 07:56:12
宝藏(2022年1期)2022-08-01 02:12:58
油气藏评价与开发(2022年3期)2022-06-23 01:55:46
环球时报(2022-04-18)2022-04-18 14:55:01
科技创新与应用(2021年7期)2021-02-04 05:12:48
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22 09:55:44
中学化学(2016年10期)2017-01-07 08:37:06
湖南城市学院学报(自然科学版)(2016年2期)2016-12-01 04:06:40
中国艺术时空(2014年2期)2014-02-28 21:32:09
